Г л а в a 1 2 МИТОХОНДРИАЛЬНАЯ ПАТОЛОГИЯ ОРГАНА ЗРЕНИЯ У ДЕТЕЙ О. В. Хлебникова
К ѴііУ'-зСЧ! іріа.іі (і;.:ѵ заболеваниям относят группу гетерогенных патологических состояний, обусловленных дефектами структуры и функции митохондрий.
Такие митохондриальные заболевания, как прогрессирующая офтальмоплегия и проірессирующая офтальмоплегия с миопатией, были известны с 1962 г.
[LuftR. et al., 1962].Этиология и генетические исследования. Особенно важным в понимании этиологии и патогенеза этой патологии является открытие в 1963 г. митохондриальной ДНК (мДНК), которая имеет двухнитчатую структуру [Nass S. et al, 1963; Taanman J.W., 1999]. Нуклеотидная последовательность мДНК состоит из 16 569 пар нуклеотидов, на которых расположено 37 генов (рис.
12.1). Каждая клетка включает от нескольких сотен до 10 000 митохондрий. Одна митохондрия содержит от 2 до 10 молекул ДНК. Комплексы дыхательной цепи митохондрий (КДЦМ) находятся пол двойным генетическим контролем; 13 полипептидов КДЦМ кодируются МДНК и около 80 полипептидов КДЦМ — ядерной ДНК. Генные мутации вызывают нарушение функции КДЦМ, что приводит к нарушению синтеза АТФ и увеличению уровня свободных радикалов в клетке (агрессивных форм кислорода). За этим следуют нарушения основных жизненно важных процессов клеточного обмена — высвобождение энергии органических веществ и аккумуляция се в виде макроэргических фосфатных соединений, происходящих в
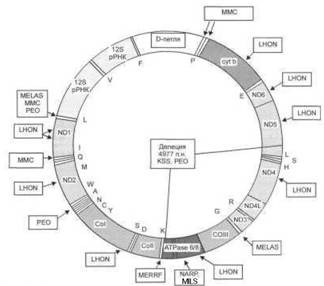
Рис. 12.1. Расположение основных мутаций мДНК.
митохондриях (метаболизм .■ і um :u і ■ P-ОКИСЛЄНИЄ, превращения в цикле К'рѵчч..;. транспорт электронов в дыхательной цепи).
Мутантная и нормальная ДНК митохондрий могут существовать в одной и той же клетке. Это состояние называется гетероплазмией [Walbace D.C., 1994J. Доля мутантной мДНК, влекущей за собой значительное снижение энергетического метаболизма клетки кч.
и >■ органа, вызывающее патологические признаки в данном органе, служит порогом фенотипической экспрессии [Graeber M.B., 1998] и является диагностическим признаком этиологии заболевания.Системные проявления. Для этой
группы заболеваний ччрчк ичч.1 выраженный внутрисемейный, внутрипо- !! \ I)! : :!,1 JI клинический полимор
физм и сочетание поражений нескольких органов и систем, которые зависят от случайного тканевого распределения мутантных митохондрий [Yang С.С. et al., 1998; Zevianl М. et al., 1992; Ozawa M. et al., 1998] и степени гетероплазмии в различных клетках и тканях. Так, например, при превышении порогового уровня мутации А3423С у больных может появиться один из синдромов: MELAS, KSS, MERRF [Mourmans J. et al., 1997; Prayson R.A. et al., 1998; Wilichowski E. et a!., 1998]. Сложность, МНОГОКОМЛО- нентность биоэнергетических процес-
сов и двойственность генетического кодирования (ядерной И ■' !' ом!і!;іі: Лея впервые описана D. Leigh в 1951 г. В настоящее время известно более 100 больных. Это гетерогенное патологическое состояние, обусловленное точковыми мутациями мДНК в нуклеотиде 8993, а также мутациями ядерной ДНК, приводящими к изменениям функции митохондрий.
у ;i;-h.oiopv> больных установлена мутация в нуклеотиде 8344, характерная для синдрома MERRF [GraeberM.B. et al., 1998]. Порог фенотипической экспрессии составляет 70—95 %.
Авторы высказывают предположение, что болезнь Лея — одна из наиболее тяжелых форм митохондриальной эниефаломиопатии — развивается в случае появления в клетках подавляющего большинства мутантной мДНК. Заболевание наследуется цитоплазматически, по аутосомно-рецессивному и Х-сцепленно-рецессивному типам.
Первые признаки заболевания могут быть обнаружены на 1—3-м годах жизни, известны и более поздние формы манифестации — с 7 лет [Ortis R.G. et al., 1993]. У ребенка появляется задержка психомоторного развития, рвота, снижение аппетита, различные нарушения мышечного тонуса, затем начинаются тонико-клонические И МИО- клонические судороги, координатор- ные расстройства, сонливость, снижение сухожильных рефлексов, нарастают симптомы пирамидной и экстрапирамидной недостаточности, нарушение глотания.
У многих детей отмечают птоз верхних век, наружную офтальмоплегию, атрофию зрительных нервов, иногда пигментную дистрофию сетчатки [Ortis R.G. et al., 1993].Течение заболевания может быть хроническим, подострым и острораз- виваюшимся с быстрым фатальным течением.
Трихополидистрофия Менкеса — болезнь «курчавых волос», описана в 1962 г. I.N. Menkes. На сегодняшний день известно около 200 больных [Николаева Е.А., 2000]. Патологический процесс развивается вследствие мутации гена, расположенного на хромосоме Xql3.3, контролирующего функцию митохондрий по транспорту меди из клетки во внеклеточное пространство.
Клинические признаки заболевания проявляются с первых недель жизни мальчиков гипотермией, недостаточной прибавкой массы тела. Затем начинаются генерализованные судороги, миоклонические подергивания лица или конечностей, мышечная гипотония сменяется спастическими парезами. Волосы у пациентов светлые, ломкие, курчавые, редкие, кожа сухая, бледная; могут быть переломы трубчатых костей.
На глазном дне у таких больных определяют атрофию зрительных нервов, микрокисты сетчатки.
Течение заболевания прогрессирующее, приводит к фатальному исходу на 1—3-М годах жизни ребенка от субдурального кровоизлияния или септического состояния. Редко наблюдаются атипичные формы болезни Менкеса с поздним началом и продолжительностью жизни до 13 лет.
Синдром D1DMOAD - симптомокомплекс, включающий диабет несахарный, диабет сахарный, оптическую атрофию, глухоту. Заболевание характеризуется прогрессирующим развитием всех перечисленных симптомов; гетерогенное, изучено плохо. Возникает при нарушении процессов репликации и транскрипции мДНК, находящихся под контролем ядерной ДНК, наследуется по аутосомно-рецессивному типу.
Синдром ARCQ — аутосомно-рецес- сивная кардиомиопатия с наружной офтальмоплегией. Заболевание обусловлено мутациями ядерного генома, приводящими к делеции мДНК. Клинико-молекулярные особенности патологии недостаточно изучены.
Таким образом, в литературе накоплен относительно небольшой опыт по изучению этиологии, патогенеза и клинических особенностей митохондриальных заболеваний глаз у детей.
Это связано с тем, что митохондриальная патология органа зрения, манифестирующая до 15 лет, как правило, входит в группу гетерогенных заболеваний с разными типами наследования, вовлечением в патологический процесс нескольких органов и систем [Вельтищев Ю.Е. и др., 1998; Краснопольская К.Д. идр., 1998; Краснопольская К.Д. и др., 1999; Николаева Е.А., 2000].Характерными признаками являют- 683
ся выраженный клинический полиморфизм и неспецифичность фенотипических проколеним Вместе с тем
нередко ,і органа
предшествуют появлению тяжелой клинической картины инвалидизи- руюших заболеваний. Это повышает роль ранней и дифференциальной диагностики митохондриальной патологии для направления на обследование в специализированные педиатрические центры.
\!л ......................................................................... патологию сле
дует дифференцировать прежде всего от заболеваний, вызванных аномалиями обмена органических кислот, которые наследуются по аутосомно-рецес- сивному типу и обусловлены мутациями ядерных ДНК.
Клинические картины заболеваний, вызванных мутациями мДНК и органическими ацидемиями, очень схожи, однако органические ацвдемии манифестируют в более раннем возрасте, птоз верхних век наблюдается только у 8 % больных и заметно усиливается после физической нагрузки, между приемами пищи; наружной офтальмоплегии, как правило, не бывает [Казанцева Л.З. и др., 1999; Краснопольская К.Д. и др., 1999].
В основном дифференциальную диагностику возможно провести на биохимическом и морфологическом уровнях.
Ранняя эти о пато генетическая диагностика митохондриальной патологии органа зрения необходима для назначения лечения, медико-генетического консультирования семьи, а в некоторых случаях и организации пренатальной диагностики.
Для лечения митохондриальной патологии у детей используют следующие препараты [Николаева Е.А., 2000]:
1) «Коэнзим Q» («убихинон») — естественный переносчик электронов от 1, 2 и 3-го комплексов дыхательной цепи; дополнительное введение его повышает активность дыхательной цепи митохондрий;
2) препараты янтарной кислоты («Янтавит», «Лимонтар») транспорти
руют І.І'.І ко 2-му комплексу
дыхательной цепи, активизируют окислительные функции митохондрий, оказывают выраженный антиоксидантный эффект;
3) «Цитохром С» — одна из ключевых субстанций дыхательной цепи митохондрий, принимает участие в переносе электронов от 3-го к 4-му комплексу;
4) «Никотинамид» выполняет функцию промежуточного переносчика восстановительных эквивалентов от различных субстратов в окислительновосстановительных процессах;
5) «Рибофлавин» — активный структурный элемент более 30 респираторных флавопротевдов.
Фдавиновые ферменты играют роль первичных дегидрогеназ при окислении сукцината и жирных кислот;6) «Тиамин», или липоевая кислота, является составной частью ко ферментов (Е| и Е2), которые участвуют в обмене углеводов, аминокислот и функции цикла Кребса;
7) і»і — кофермент ряда ко-
карбоксилаз, участвующих в метаболизме пировиноградной кислоты;
8) «Карнитин», его главная функция — перенос жирных кислот через митохондриальную мембрану — влияет на энергетический метаболизм внутри митохондрий;
9) «Димефосфон» активизирует тканевое дыхание и стабилизирует состояние клеточных мембран;
10) «Токоферол» (витамин Е) — жирорастворимый витамин с выраженным антиоксидантным действием, нормализует структуру и проницаемость биологических мембран, оказывает положительное действие на интенсивность клеточного дыхания, активизирует ферменты и коферменты, содержащие сульф гидр ильные группы;
I I 1 витамин С участвует во многих окислительно- восстановительных процессах благодаря способности обратимо окисляться в дегидроаскорбиновую кислоту. Кроме того, он действует как переносчик электронов на уровнях 3 и 4 дыхательной цепи.
Однако объективного клинического подтверждения длительного улучшения зрительных функций и замедления процесса заболевания при динамическом наблюдении не представ-
Поиски патогенетически обусловленной терапии продолжаются
Хирургическое лечение птоза и косоглазия может привести к осложнениям [Ehner R, 1999], авторы наблюдали появление стойкой диплопии после удаления катаракты у больных с митохондриальной патологией
Еще по теме Г л а в a 1 2 МИТОХОНДРИАЛЬНАЯ ПАТОЛОГИЯ ОРГАНА ЗРЕНИЯ У ДЕТЕЙ О. В. Хлебникова:
- 2.3. особенности реабилитации инвалидов с патологией органа зрения
- Особенности коррекции при различных видах патологии органа зрения
- ЧАСТЬ IIЗАБОЛЕВАНИЯ ПЕРЕДНЕГО ОТДЕЛА ОРГАНА ЗРЕНИЯ У ДЕТЕЙ
- Состояние органа зрения
- Наружный осмотр органа зрения
- Ожоги органа зрения (код Т26)
- Особенности медико-социальной экспертизы больных с последствиями травм органа зрения
- Применение электрофизиологических методов исследования при повреждении органа зрения
- Проблема контакта органа зрения и объекта
- Экспертная оценка лиц с заболеваниями органа зрения