О применении потенциала Гупта для описания межмолекулярного взаимодействия в металлических системах
В последние годы достигнут значительный прогресс как в теории, так и в развитии соответствующих численных алгоритмов в физике конденсированных сред. Одним из направлений компьютерного моделирования свойств металлов, в том числе исследования фазового перехода кристалл - расплав, является применение метода Монте-Карло (MK) с полуэмпирическими потенциалами, параметры которых подбираются по экспериментальным данным, как правило относящимся к массивной фазе.
При исследовании методами компьютерного моделирования систем, содержащих нейтральные атомы металлов и металлические наночастицы, в частности для определения термодинамических характеристик, необходимо использовать потенциал, адекватно описывающий их взаимодействие. К настоящему времени выполнено достаточно большое число работ посвященных решению задачи восстановления металлического потенциала [76, 81, 92]. Обычно для этого используются экспериментальные данные по энергии когезии атомов. В настоящее время в расчетах свойств металлических нанокластеров наиболее часто используется потенциал Гупта и его модификации [62, 81, 89, 92, 168]. Подобно другим металлическим потенциалам, потенциал Гупта восстановлен в рамках теории функционала плотности по энергии когезии атомов металлического тела и описывает взаимодействие в терминах локальной электронной плотности. Отметим также, что в силу нелинейности и многочастичности потенциала Гупта проведение прямых вычислений с его использованием требует слишком больших затрат машинного времени.
Для построения атомистической модели наночастиц диаметра Dбыл использован следующий алгоритм:
• сначала строится гранецентрированная кубическая решетка большого размера с заданной длиной ребра элементарной ячейки и углом между ребрами 90°;
• далее выбираются координаты тех атомов, которые целиком принадлежат сфере диаметра D.
Такой алгоритм неизбежно приводит к появлению огранки наночастиц, в частности, можно привести пример, когда наблюдаемые наночастицы золота также обладают огранкой [169].
Значение полной потенциальной энергии наночастиц, содержащей N атомов металла в случае использования потенциала Гупта, дается выражением:
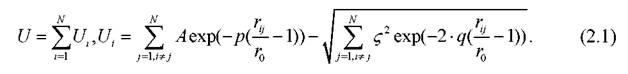
Как и любой эмпирический потенциал, потенциал Гупта получен путем обработки экспериментальных данных с приведением к удобной математической форме записи. Первый член формулы (2.1) является парным потенциалом отталкивания (описывается суммой Борна - Майера ион-ионного отталкивания, полученного из приращения кинетической энергии электронов проводимости, ограниченных двумя приближающимися ионами [168]), второе слагаемое - потенциалом притяжения. Как видно из (2.1), именно второе слагаемое обусловливает неаддитивность потенциала. Отметим, что вклад, отвечающий притяжению, представляет собой энергию, пропорциональную второму моменту электронной плотности. Это слагаемое квантового происхождения, включающее как раз многочастичное суммирование. В формуле (2.1) rij- расстояние между атомами z и у в кластере, параметр А - экспериментальное значение энергии когезии, r0- параметр кристаллической решётки, p,q - значения упругих постоянных кристаллической структуры при T = 0 К, относящиеся к массивному образцу. Для атомов золота, меди, алюминия и кобальта параметры потенциала взяты из [81] (см. Таблицу 1). Отметим также короткодействующий характер потенциала Гупта, данное свойство, в том числе, может обусловливать
специфическое поведение размерных зависимостей термодинамических и структурных характеристик, рассмотренных в главе 3. Взаимодействие потенциалов двух наночастиц на коротких дистанциях определяется вкладами атомов, принадлежащих только «контактной зоне». Таким образом, в частности, в [170] для описания взаимодействия получено парное выражение для аппроксимации потенциала Гупта:
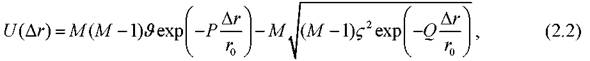
где M - среднее число атомов, принадлежащих контактной зоне, Ar - расстояние между двумя ближайшими точками, лежащими на поверхности двух наночастиц.
Таблица 1. Параметры потенциала Гупта.