Обзор вычислительных методов, используемых при моделировании
В последние годы достигнут значительный прогресс как в теории, так и в развитии численных алгоритмов в физике конденсированных сред. Здесь мы рассмотрим основные численные методы, используемые в моделировании наноструктур.
Следует отметить, что каждый раз необходимо делать выбор из большого числа методов. Например, при моделировании больших наночастиц нелегко применять квантово-механические методы и поэтому приходится использовать классическое описание. Далее кратко опишем некоторые методы, их преимущества и недостатки.Метод Борна - Оппенгеймера
Метод Борна - Оппенгеймера [38] является разновидностью адиабатического приближения уравнения Шрёдингера в квантовой механике, метод анализа молекулярных систем, заключающийся в том, что в системе выделяют и раздельно описывают ядра атомов и электроны, для которых характерные времена изменения состояния сильно различаются. Масса ядра значительно превышает массу электрона, вследствие чего скорость движения ядер мала по отношению к скорости движения электронов. В результате медленно движущиеся ядра образуют электростатическое поле, в котором с намного большей скоростью движутся электроны, успевающие мгновенно подстроиться к любому изменению координат ядер. Поэтому в приближении считают ядра фиксированными и рассматривают только движение электронов. На языке квантовой механики это эквивалентно допущению, что полная волновая функция молекулы может быть выражена в виде произведения электронной и ядерной функций Ψ(7ζr) = Ψπuc(A)∙Ψe∕(7ζr), где г - координаты электронов, a R- ядер. Приближение Борна - Оппенгеймера является важным для квантовой химии. В этом приближении полная энергия представляет собой сумму электронной энергии, вычисленной при фиксированной конфигурации ядер, и колебательно-
32 вращательной энергии ядер: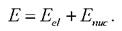 Ясно, что этот метод имеет
Ясно, что этот метод имеет
ограничения на лёгкие ядра химических элементов.
Метод Хартри-Фока
Метод Хартри - Фока [39-41] состоит из нескольких стадий. На первом этапе решается задача о движении электрона в определённом модельном потенциале, который должен как можно лучше отображать взаимодействие выбранного электрона с ядрами атомов и другими электронами. Найденные волновые функции используются для того, чтобы определить взаимодействие электрона с другими электронами и ядрами, уточняя потенциал. В дальнейшем опять решается задача нахождения волновых функций электрона для нового потенциала и нахождения из него следующего, более точного. Процедура продолжается до достижения сходимости.
Волновая функция многоэлектронной системы выбирается в виде детерминанта Слэтера. Уравнения Хартри - Фока являют собой одноэлектронные уравнения типа уравнения Шрёдингера, которым соответствуют орбитали, отвечающие минимальным значениям энергии молекулярной системы. В простейшем случае уравнения Хартри - Фока имеют вид
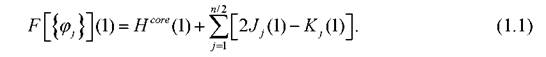
Здесь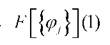 - фокиан, являющийся оператором Гамильтона для одного
- фокиан, являющийся оператором Гамильтона для одного
электрона, находящегося в самосогласованном поле. Фокиан состоит из суммы одноэлектронного оператора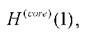 равного сумме оператора кинетической
равного сумме оператора кинетической
энергии электрона и оператора потенциальной энергии его взаимодействия со всеми ядрами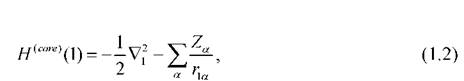
и суммы операторов 2J (I)-X (1), определяющих взаимодействие рассматриваемого электрона с усреднённым полем остальных электронов.
Действие двух последних операторов на орбиталь φопределяется следующими соотношениями: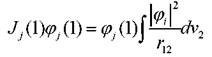 - оператор Кулона, учитывающий
- оператор Кулона, учитывающий
взаимодействие с орбиталью у-го электрона,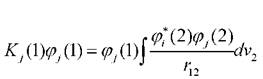 -
-
обменный оператор.
Основным недостатком метода является то, что он не учитывает корреляционную энергию для электронов.Кроме того, существуют модификации этого метода в виде методов Хартри - Фока - Боголюбова, Хартри - Фока - Дирака, Хартри - Фока - Слейтера и Хартри - Фока - Рутана [42-44].
Метод функционала плотности
Методу теории функционала плотности предшествовала модель Томаса - Ферми, развитая Л. Томасом и Э. Ферми в 1927 году. Они рассчитали энергию атома как сумму его кинетической энергии, представленной в виде функционала электронной плотности и потенциальной энергии взаимодействия электронов с ядром и друг с другом; энергия взаимодействия также была выражена через электронную плотность [45-49].
Метод функционала плотности (МФП) является одним из наиболее перспективных методов изучения структурных и, как следствие, физических и физико-химических свойств жидкостей и твердых тел. Данный метод применяется для изучения вещества как на микроскопическом, так и на мезоскопическом уровнях. В настоящее время мезоскопический вариант МФП (метода функционала атомной плотности - МФАП) гораздо менее известен, чем метод функционала электронной плотности. МФП на электронном уровне позволил решить ряд важных задач теории конденсированного состояния, в частности определение размерных эффектов в металлических кластерах нанообъектов [50]. В работе Т.В. Быкова и А.К. Щекина МФАП был применен к проблеме нахождения распределения плотности в малой капле и ее
34 поверхностного натяжения [51], кроме того, мезоскопический вариант МФ АП применялся к проблеме гетерогенной нуклеации на микроскопических смачиваемых частицах [51,52]. В диссертационной работе [53] МФ АП был применен к исследованию структурных и термодинамических характеристик конденсированных пленок на поверхности твердого тела.
Несмотря на заметную роль, которую модель Томаса - Ферми сыграла в развитии квантовой механики, её точность была недостаточной, поскольку не учитывалось обменное взаимодействие, в отличие, например, от метода Хартри - Фока.
В 1928 году П. Дирак уточнил функционал энергии в модели Томаса - Ферми, добавив к нему слагаемое, описывающее обменное взаимодействие (это слагаемое по структуре также имело вид функционала электронной плотности). Несмотря на это, для ряда применений модель Томаса - Ферми - Дирака не давала удовлетворительного результата. Основным источником погрешности являлось выражение кинетической энергии, приводящее к погрешности в вычислении обменной энергии. Кроме того, не учитывалась энергия электронной корреляции.Хотя теория функционала плотности и базируется на ставшей классической модели Томаса - Ферми, надёжное теоретическое обоснование под нее было подведено только с формулировкой теорем Хоэнберга - Кона.
Первая теорема утверждает, что существует взаимно однозначное соответствие между плотностью основного состояния электронной подсистемы, находящейся во внешнем потенциале атомных ядер, и самим потенциалом ядер. Первая теорема является теоремой существования и не дает метода построения такого соответствия.
Вторая теорема представляет собой вариационный принцип квантовой механики, сформулированный для функционала плотности, и утверждает, что энергия электронной подсистемы, записанная как функционал электронной плотности, имеет минимум, равный энергии основного состояния: 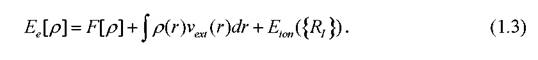
Здесь vext(r) - потенциальная энергия взаимодействия электронного газа с внешней средой,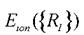 - энергия взаимодействия ионов между собой,
- энергия взаимодействия ионов между собой,
зафиксированных в точках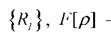 - универсальный функционал плотности
- универсальный функционал плотности
р, не зависящий от vext(r).К сожалению, точная форма функционала F[p}не известна, однако существуют разумные приближения, такие, как приближения локальной плотности (LDA), обобщённая градиентная аппроксимация (GDA) и гибридные методы [46-48].
Первоначально теоремы Хоэнберга - Кона были сформулированы только для основного состояния электронной подсистемы в отсутствие магнитного ПОЛЯ. Они могут быть обобщены путём введения зависимости от времени, что позволяет использовать этот формализм для расчета состояний возбуждённых электронов [49].
Традиционные методы определения электронной структуры, в частности метод Хартри - Фока и производные от него, описывают систему с помощью многоэлектронной волновой функции. Основная цель теории функционала плотности - при описании электронной подсистемы заменить многоэлектронную волновую функцию электронной плотностью. Это ведет к существенному упрощению задачи, поскольку многоэлектронная волновая функция зависит от ЗА переменных - по три пространственные координаты на каждый из N электронов, в то время как плотность - функция лишь трёх пространственных координат.
Как правило, метод теории функционала плотности используется совместно с формализмом Кона - Шэма, в рамках которого трудноразрешимая задача об описании нескольких взаимодействующих электронов в статическом внешнем поле (атомных ядер) сводится к более простой задаче о независимых электронах, которые движутся в некотором эффективном потенциале. Этот эффективный потенциал включает в себя статический потенциал атомных ядер, а также учитывает кулоновские эффекты, в частности обменное взаимодействие и электронную корреляцию.
Описание двух последних взаимодействий и представляет собой основную сложность метода теории функционала плотности в формулировке Кона-Шэма. Простейшим приближением здесь является приближение локальной плотности, основанное на точном расчёте обменной энергии для пространственно однородного электронного газа, который может быть выполнен в рамках модели Томаса - Ферми и из которого можно получить также и корреляционную энергию электронного газа.
Метод теории функционала плотности широко применяется для расчётов в физике твёрдого тела с 70-х годов XX века. В ряде случаев даже использование простого приближения локальной плотности дает удовлетворительные результаты, соответствующие экспериментальным данным, причём вычислительная сложность метода невысока относительно других подходов к проблеме многих частиц в квантовой механике.
Тем не менее долгое время метод был недостаточно точен для расчётов в области квантовой химии, пока в 90-х годах XX века не произошёл заметный сдвиг в описании обменного и корреляционного взаимодействий. В настоящее время метод теории функционала плотности является главным подходом в обеих областях. Впрочем, несмотря на прогресс в теории, все ещё имеются проблемы в приложении метода к описанию межмолекулярных сил, в особенности Ван-дер-Ваальсовых сил и дисперсионного взаимодействия, а также в расчётах ширины запрещённой зоны в полупроводниках. Сложности с расчётом дисперсионного взаимодействия в рамках теории функционала плотности (которые возникают, как минимум, в том случае, когда этот метод не дополняется другими) делают метод теории функционала плотности малопригодным для систем, в которых дисперсионные силы являются преобладающими (например, при рассмотрении взаимодействия между атомами благородных газов), или систем, в которых дисперсионные силы имеют тот же порядок, что и другие взаимодействия (например, в органических молекулах). Решение этой проблемы является предметом современных исследований.
На практике метод Кона-Шэма может быть применён несколькими различными способами, в зависимости от цели исследования. В расчётах для физики твёрдого тела до сих пор широко используется приближение локальной плотности, вкупе с базисом плоских волн. Для расчётов электронной структуры молекул требуются более сложные выражения для функционалов. Так, большое число приближенных функционалов для расчёта обменно-корреляционного взаимодействия было развито для задач химии. Некоторые из них противоречат приближению пространственно однородного электронного газа, но тем не менее в пределе при переходе к электронному газу должны сводиться к приближению локальной плотности.
Для расчётов физических задач наиболее часто применяется, по-видимому, уточнённая обменная модель Perdew-Burke-Emzerhof, однако известно, что она приводит к ошибкам в калориметрических параметрах, будучи приложенной к расчётам молекул в газовой фазе.
В расчётах квантовой химии одним из распространённых является вид обменного функционала, называемый BLYP (Becke, Lee, Yang, Parr). Еще более широко распространено приближение B3LYP, основанное на гибридном функционале, в котором обменная энергия рассчитывается с привлечением точного результата, полученного методом Хартри-Фока.
В целом текущее состояние метода теории функционала плотности таково, что невозможно оценить погрешность расчёта, не сравнивая его результаты с другими подходами или с результатами экспериментов.
Классические методы (метод Монте-Карло и метод молекулярной динамики)
К числу классических методов моделирования относятся молекулярная динамика (МД) и метод Монте-Карло (MK) [54-56]. Метод MK дает информацию о конфигурационных характеристиках системы (хотя имеется также и их динамическая интерпретация [56]), в отличие от метода МД, дающего также и динамические характеристики. Метод МД дает информацию о временной зависимости и величине импульса и координат частиц. Путем выбора
38 соответствующего ансамбля, такого, как канонический, метод MK позволяет вычислять наблюдаемые переменные при фиксированных числе частиц, объеме и температуре. Большое преимущество метода MK состоит в том, что необходимое множество ансамблей легко реализуется, а сами ансамбли можно также изменять во время моделирования.
Об описании взаимодействия при моделировании
Во многих случаях невозможно использовать методы электронной структуры. В этом случае выбор классических методов (или, как их ещё называют, «атомистических» методов) часто является наиболее оптимальным выбором. Они широко используются для исследования морфологических и эволюционных явлений. Классические методы не подходят для описания квантово-механических свойств, таких, как электронная структура или спин. Однако широкий выбор различных потенциалов даёт возможность хорошего описания химических и термодинамических свойств. Широкий обзор различных потенциалов приведён в [57].
Существует две известные модели, описывающие атомное взаимодействие в металлических наночастицах, - модель погружённого атома (EAM) [58,59] и модель Финнеса - Синклера [60]. Оба потенциала получаются из теории функционала плотности [45] и описывают связь атомов в терминах электронной плотности. Для однокомпонентных систем эти потенциалы оказываются одинаковыми. Однако существует тонкое различие при использовании их для моделирования сплавов. Общая форма этих потенциалов такая:
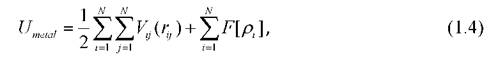
где F[pJ - функционал, описывающий энергию погружённого атома в зависимости от плотности pi, которая определяется соотношением 
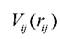 - потенциальная энергия парного взаимодействия, включающего в себя отталкивающее электростатическое и перекрывающее взаимодействие. Метод EAM использует многочастичный потенциал, и, поскольку плотность электронного облака - это сумма вклада от большого количества атомов, на практике для уменьшения сложности и соответственно времени расчетов часто ограничивают количество соседей так называемым «радиусом обрезания». Приведём конкретные примеры потенциалов, полученных в рамках этой модели. Потенциал Финнеса-Синклера
- потенциальная энергия парного взаимодействия, включающего в себя отталкивающее электростатическое и перекрывающее взаимодействие. Метод EAM использует многочастичный потенциал, и, поскольку плотность электронного облака - это сумма вклада от большого количества атомов, на практике для уменьшения сложности и соответственно времени расчетов часто ограничивают количество соседей так называемым «радиусом обрезания». Приведём конкретные примеры потенциалов, полученных в рамках этой модели. Потенциал Финнеса-Синклера
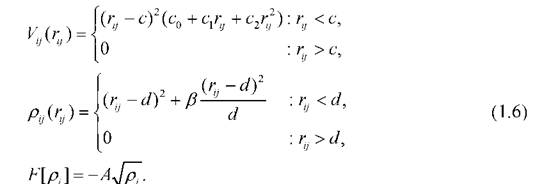
Здесь с, d, c0, c1, с2, Д А - параметры, зависящие от свойств материала. Потенциал Саттон-Чена
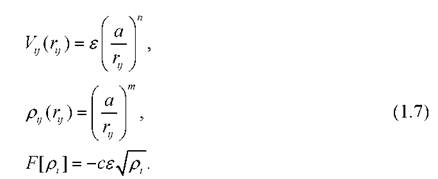
Здесь ε,a,c,m,n - параметры, зависящие от свойств материала.
Потенциал Гупта
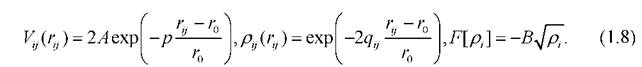
Здесь A,p,r0,q В - параметры, зависящие от свойств материала. Потенциал Гупта был успешно апробирован в работах сотрудников кафедры общей физики Тверского государственного университета [61-67].
Молекулярная динамика
Метод молекулярной динамики основан на численном решении системы уравнений Ньютона и позволяет описать поведение системы с течением времени. Известно, что состояние механической системы описывается 6N-мерным вектором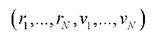 положений частиц и скоростей. Эволюция этого
положений частиц и скоростей. Эволюция этого
вектора в фазовом пространстве описывается уравнениями Ньютона:

Для решения уравнений (1.4) требуется задание некоторого начального условия
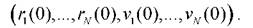
Существует множество численных методов для решения системы (1.4), среди них часто используется алгоритм Верле [68,69]. В этом алгоритме предполагается, что движение частицы в течение времени Mявляется прямолинейным и равномерным, поэтому изменение положения и скорости на каждой итерации можно вычислять так: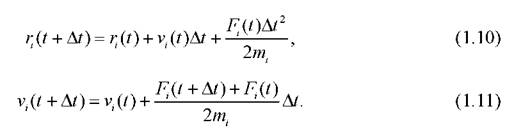
Одной из наиболее существенных проблем процедуры интегрирования является выбор шага. При большом шаге погрешности интегрирования могут быть значительными, что приведёт к нестабильности траектории. При малом шаге существенно увеличивается время расчёта. В уравнениях движения, описывающих изменения по различным степеням свободы, временные характеристики существенно отличаются друг от друга. Для достаточно точного вычисления решения по быстрым и медленным переменным шаги интегрирования по ним могут различаться. По быстрым переменным может быть выбран значительно больший шаг. В методе Верле шаг интегрирования берётся единым, оптимальным считается шаг 1-1,5 фс, что является примерно десятой частью периода самых быстрых молекулярных колебаний. Начальные скорости
41 атомов выбираются с помощью генератора случайных чисел в соответствии с распределением Максвелла при заданной температуре.
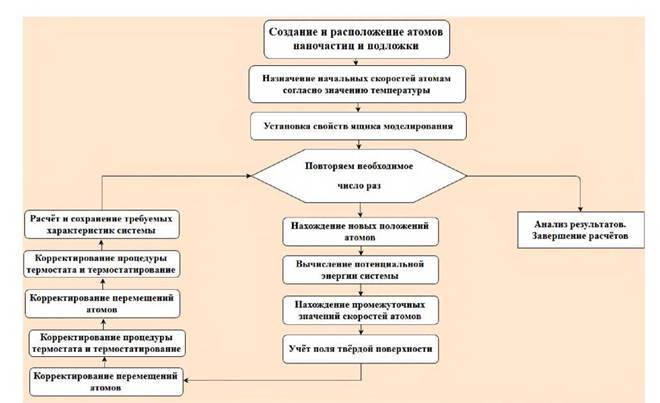
Рис. 1. Блок-схема работы программы молекулярной динамики [70].
Основной задачей статистической теории является вычисление средних значений различных физических величин, которые представляют собой функциии микросостояния системы. Рассмотрим, как эта проблема решается в рамках молекулярной динамики.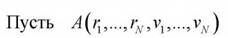 - некоторая физическая
- некоторая физическая
величина, являющаяся функцией микропараметров. Тогда среднее значение этой величины определяется путем усреднения множества «мгновенных» значений 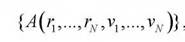 , наблюдаемых в последовательные моменты времени tна достаточно большом интервале T:
, наблюдаемых в последовательные моменты времени tна достаточно большом интервале T: 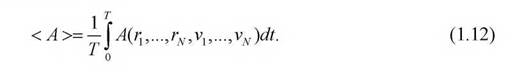
Такое усреднение по времени исходит из того, что нам известны законы движения частиц системы. На рис. 1. приведена блок-схема алгоритма, применяемая при МД-моделировании.
Метод Монте-Карло
Другой путь вычисления средних значений физических величин, являющихся параметрами системы, был намечен Больцманом, а затем развит Гиббсом. Здесь рассматривают множество состояний системы, а затем для каждого состояния вычисляют значение физической величины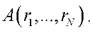 После
После
этого производят усреднение по всем значениям. Такой подход называется усреднением по ансамблю. Вероятность возникновения того или иного состояния пропорциональна его статистическому весу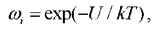 где U -
где U -
потенциальная энергия данной конфигурации, к - константа Больцмана, T - абсолютная температура. Среднее значение физической величины √4(r1,...,rv) вычисляется по формуле
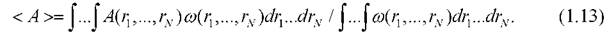
При оценке интеграла (113) значения наблюдаемой величины  в принципе можно выбирать, задавая случайным и равновероятным способом координаты частиц. Подобная вычислительная процедура представляет собой простейший вариант метода статистических испытаний Монте-Карло - метод независимых проверок. В этом случае интеграл (1.13) можно преобразовать к виду
в принципе можно выбирать, задавая случайным и равновероятным способом координаты частиц. Подобная вычислительная процедура представляет собой простейший вариант метода статистических испытаний Монте-Карло - метод независимых проверок. В этом случае интеграл (1.13) можно преобразовать к виду
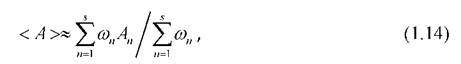
выбранных конфигураций системы. Метод независимых выборок (схема Метрополиса) сводится к следующей последовательности действий:
1. Случайным образом генерируется конфигурация систем из заданного числа частиц Nпри заданной их средней плотности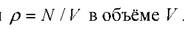
2. Вычисляется потенциальная энергия Unданной конфигурации п .
3. Измеряется требуемая характеристика системы, которая вносит свой вклад в общую статистику (среднюю величину), пропорционально статистическому весу
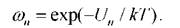
4. Шаги 1-3 повторяются большое число раз 5, а требуемые статистические средние наблюдаемых величин находятся из соотношения (1.14).
Обычно статистический вес ωимеет очень широкие пределы изменения. Поэтому расчёт по формуле (1.14) приводит к большой дисперсии усредняемых величин. Для понижения этой дисперсии используется приём, который называется существенной выборкой. Если при этом в качестве весовой функции рассматривать статистический вес данной конфигурации, т.е. проводить выборку состояний системы не равновероятным образом, а с вероятностью, пропорциональной ωn,то выражение (1.14) сведётся к виду

Впервые схема существенной выборки при компьютерном моделировании была использована в знаменитой работе Метрополиса, супругов Розенблатов и супругов Теллеров [71]. В этом методе с помощью генератора случайных чисел разыгрываются переходы между конфигурациями системы при небольших изменениях последовательных конфигураций. Этот алгоритм выглядит так:
1. На начальном этапе следует случайным образом создать систему, состоящую из требуемого числа частиц N, при плотности р. Вычисляется значение потенциальной энергии Un.
2. Случайным образом выбирается одна из частиц і с координатами ri =(xi,yi,zi').
3. Генерируются три случайных числе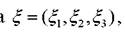 равномерно распределённых
равномерно распределённых
на интервале [0,1]. Делается попытка переместить выбранную частицу ів новую
44 позицию с координатами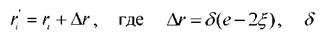 - малое
- малое
положительное число, е = (1,1,1).
4. Вычисляется новая потенциальная энергия Uсистемы. Если U Un, то частица переходит в новое положение с вероятностью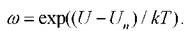 Для этого генерируется
Для этого генерируется
ещё одно случайное число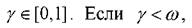 то переход принимается, в
то переход принимается, в
противном случае частица возвращается на прежнее место, при этом полученная конфигурация всё равно участвует в общей статистике.
5. Цикл повторяется, начиная с пункта 2.
Отдельная попытка перемещения частицы (удачная или неудачная) называется микрошагом. После того как число выполненных микрошагов станет кратным числу частиц N, измеряются требуемые характеристики системы. Ццкл из Nмикрошагов называется макрошагом. Один макрошаг рассматривается в качестве условной единицы времени τэволюции системы. Полученные значения накапливаются и вносят равный вклад в статистику. Поэтому после s-кратного повторения шагов 2-4 требуемые средние величины определяются как простые средние арифметические по формуле (Е15).
Более подробное описание метода Монте-Карло можно найти, в частности, в монографии [56].
1.3.
Еще по теме Обзор вычислительных методов, используемых при моделировании:
- 3.1. Обзор существующих методов и средств моделирования
- Методы используемыми при формировании и развитии организационной культуры
- Результаты рассмотрения поведения наночастиц при фазовом переходе 1 рода, полученные классическими методами моделирования
- Глава 3. О результатах компьютерного эксперимента по моделированию термодинамических и структурных характеристик при фазовом переходе первого рода для нанокластеров металлов методом Монте-Карло
- А. И. Анциперова, В. Г. Нестолий. Моделирование примерных форм процессуальных документов, используемых при предъявлении иска и в исполнительном производстве : учебное пособие / А. И. Анциперова, В. Г. Нестолий ; под ред. В. Г. Нестолия; РПА Минюста России, Иркут, юрид. ин-т (филиал). — Иркутск : РПА Минюста России, 2013, 2013
- Вопрос 2 Методы исследования, используемые в этнопсихологии.
- 19. Научные методы, используемые в экономической географии.
- Методы нейросетевого управления, используемые в биотехнических системах
- § 2.8. Методы климатического ГИС-моделирования.
- 3.3. Методы моделирования и количественного анализа для решения управленческих проблем
- 6. Метод моделирования в экономике. Кривая производственных возможностей.
- 4.2. Моделирование систем массового обслуживания с использованием метода Монте-Карло
- Моделирование ошибок при вычислении местоположения поданным одометрии
- Автотранспортные Cpedcmeat используемые при ведении наружного наблюдения
- Метод корреляционного моделирования
- 3. Методы, используемые для обращения к аудитории
- 3.1 Оборудование, используемое при исследовании тягового сопротивления
- Обзор методов