Х-сцепленный врожденный (ювенильный) ретиношизис
Х-сцеплснныЙ врожденный (ювенильный) ретиношизис — сцепленная с полом двусторонняя врожденная витрео- ретинальная дегенерация. Подавляю-
большинство таких больных — лица мужского пола.
Частота заболевания в популяции — 1 : 10 000 [Bergen А.А.В. et al., І995|.Заболевание впервые описано J. Haas и К. Stabsarzt (1898), которые наблюдали «радиальную кистозную ма- кулопатию» у двух мужчин и высказали предположение о том, что заболевание имеет воспалительную природу. Через 15 лет Н.Е. Pagenstecher опубликовал сообщение о семье, у нескольких членов которой был выявлен рети- ношизис. В его публикации был показан сцепленный с палом тип нвеледо- птіі-ш заболевания [Pagenstecher H.E., 1913]. Несколько позже J. Anderson (1932) и Е. Thomson (1938) описали изменения на периферии сетчатки у пациентов с ретин ОШ из ис ом.
A. Sorsby и соавт. (1951Ц дали описание 8 мужчин с ретиношизисом из 3 поколений одной семьи, включающей более 80 родственников, сделав вывод о сцепленным с полом рецессивном характере наследования заболевания. Поданеє появились многочисленные сообщения о семьях, в которых и 4—7 поколениях были обнаружены больные с Х-сцепленным врожденным ретиношизисом [ Duke- Elder S., DobreeJ.H., 1967; Huopaniemi L. et al., 1999; Mashima Y. et al., 1999; Mendoza-Lon don о R. et al., 1999].
Генетика и патогенез. Заболевание передается через Х-хромосому, болеют преимущественно лица мужского пола. Женщины обычно клинически здоровы и являются носителями патологической X-хромосомы. Их СЫНОВЬЯ имеют 50 % шанс унаследовать заболевание, а дочери — 50 % вероятность стать носителями. Н. Forsius и соавт. (1969) обнаружили случай гомозиготного поражения женшины, у отца которой был Х-сцеплснный врожденный ретиношизис, а мать, являющаяся его двоюродной сестрой, оказалась носителем патологического гена. R. Mendo- za-Londono и соавт. (1999) описали большую колумбийскую семью, у членов которой наблюдался Х-сцеплен- ный врожденный ретиношизис.
Характерные клинические и электрофизиологические изменения выявлены у 27 мужчин и 3 женщин. Тяжесть функциональных и клинических проявлений заболеваний у мужчин и женщин была одинаковой [Mendoza-Lon do- no R. et al., 1999].Обнаружены другие варианты наследования врожденного ретиношизи- са, в частности аутос ом но-рецессивный и аутосомно-доминантный [Lewis R.A. et al., 1977; Yassur Y. et al., 1982; Shimazaki J., Matsuhashi M., 1987]. Эти заболевания развиваются у лиц не только мужского, но и женского пола.
Ген RS1, ответственный за развитие ретиношизиса, был картирован в дистальной части короткого плеча Х-хро- м ос омы в интервале Хр22.1 — р22.2 |AlitaloT. et al., 1987]. Сцепление гена RS! с двумя полиморфными маркерными генами — DXS43 и DZS41 в Хр22.1 — р22.2 было установлено в исследованиях A. Gal (1988) и N. Dahl и соавт. (1988). G. Gellert и соавт. (1988) при проведении молекулярных генетических исследований у членов 6 семей с Х-сцепленным врожденным ретиношизисом обнаружили, что ген RS1 более тесно сцеплен с маркерным геном DXS9. Авторы предположили следующий порядок расположения гена и маркеров в Хр21.1 — р22.2: RS1 — DXS9 - DXS43 - DXS16 - DXS41.
Ген RS1 образован шестью j'н.і ми, кодирующими 224 аминокислоты белка-предшественника. который включает лидерный пептид, состоящий из 23 аминокислотных остатков, и вы со ко консервативныйдискообраз- ные- исследования 'других I балков(с дискообразными мотивами показали, что этот домен играет важную роль в фосфолипидном связывании и клеточной адгезии (Gehrig A.E. et al., 1999]. Подобные функциональные свойства могут быть присущи и белку RS1, что коррелирует с патоморфологическими изменениями при ретиношизисе. Мутации, вызывающие изменения структуры белка или его преждевременную
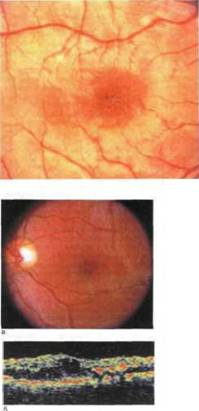
Рис. 9.2. Х-сцепленный врожденный (ювенильный) ретиношизис а — офтальмоскопическая картина микро, кистозные нагошения, приводящие к формированию углубления в фовеоле (пончикоподобные изменения); б — оптическая когерентная томограмма: множественные микрокисты в
Рис.
9.1. Х-сцепленный врожденный (ювенильный) ретиношизис. Фовеолярные микрокисты, образующие фигуру спиц в колесе.деградацию в момент процессинга, приводят к растеплению сетчатки между слоем нервных волокон и внутренней пограничной мембраной [Yanoff М. et al., 1968]. Другой м орф ол о- гический дефект — дегенеративные изменения клеток Мюллера, обусловленные нарушением транспорта ионов К+ [Мосин И.М. и др., 2001; Condon G.P. et al., 1986].
Установлено около 100 различных мутаций, приводящих к нарушениям функциональной активности белка RS1 и ответственных за развитие Х-сцепленного врожденного ретиношизиса. Они включают внутригенные микроделе- ции (7 % случаев), нонсенс-мутации (6 %), миссенс-мутации (75 %), мутации в сайте сплайсинга (6 %), а также небольшие вставки и делеции, приводящие к сдвигу рамки считывания (6 %) [Multicenter study, 1998; Huopa- niemi L. et al., 1999; Mashima Y. et al., 1999; Mendoza-Londono R. et a!., 1999; Shastry B.S. et al., 1999].
Экзоны 1—3 содержат небольшое количество мутаций, приводящих, как правило, к образованию усеченных транслянтов. Это свидетельствует о незначительной функциональной роли этого сегмента в белке [Rodriguez I.R. el al., 1998]. Экзоны 4—6, кодирующие дисковидный домен, содержат наибольшее число мутаций, преимущественно миссенс-мутаций [Huopanie- mi L. et al., 1999; Mashima Y. et al., 1999; Shastry B.S. et al., 1999]. При их анализе выявлено преобладание мутации Argl97Cys в сайте, имеющем фатальное значение для образования третичной структуры и, следовательно, для функционирования белка [Mashi- ma Y. et Y 1999; Shaslry B.S. et al.. 1999]. Часто наблюдаются также мутации некоторых аминокислот, которые, возможно, участвуют в RS/специфичных белок-белковых взаимодействиях на поверхности мембран (Multicenter study, 1998].
R. Mendoza-Londono и соавт. (1999) выявили делецию одного основания (G) в позиции 639 (639 del G), вызывающую сдвиг рамки считывания при трансляции. В результате этого синтезируется белок с резко увеличенным дисковидным доменом, содержащий 235 аминокислот вместо 224 и превосходящий нормальный по длине.
Несмотря на гетерогенность мутаций, вызывающих ретиношизис, патоморфологические механизмы заболевания у пациентов из разных семей в значительной степени схожи, хотя отмечаются выраженные различия в клинической картине и характере течения. Не выявлено фенотипа врожденного ретиношизиса, который был бы характерен для больных женского пола [Muiticenter study, 1998; Mendoza-Londono R. et al., 1999].
Г[7]:.>ф|.і;!кі!!і5;. у детей с Х-сцепленным врожденным ретиношизисом обычно бывает гиперметропия, часто сочетающаяся с астигматизмом [Мосин И.М., 2000; HottaY. et al., 2001]. Гиперметропия у больных с Х-сцепленным врожденным ретиношизисом вызвана укорочением аксиальной длины глазного яблока [Мосин И.М. и др., 2001; Ка- to К. et al., 2001].
Системные проявления. Фовеолярные изменения, обусловленные наличием микрокист, диагностируют 98—100 % больных уже в раннем возрасте. При офтальмоскопии в макуле выявляются радиальные складки, называемые симптомом «спиц в колесе» (рис. 9.1). Позднее в центре формируется углубление (рис. 9.2) с приподнятыми краями (фигура, напоминающая пончик), а в исходе заболевания — участки хориоретинальной атрофии с отложениями пигмента и твердого экссудата (рис. 9.3) [Мосин И.М., 2000; Захарова Г.Ю., Зуева М.В., 2001;

Мосин И.М. и др., 2001; Harris J.S., Yeung J.W.S., 1976; Tasman W., 1999]. Иногда в заднем полюсе фиксируют точечные белые очажки, напоминающие изменения при абиотрофии fundus albipunctatus [van Schooneveld M.J., Miyake Y., 1994; HottaY. et al., 2001].
Периферические нарушения, представляющие обширные участки расщепления сетчатки на уровне слоя нервных волокон и локализующиеся преимущественно в нижне наружном квадранте, выявляют у 60—70 % пациентов (рис. 9.4). Часто обнаруживают дегенеративные изменения сетчатки в виде рассеянных очажков гиперпигментации и золотисто-серебристых у участков, напоминающих поверхность битого металла (рис. 9.5), а также аномальные сосуды с повышенной проницаемостью, образующие белые древовидные структуры.
В стекловидном теле определяются тяжи, аваскулярные или васкулярные мембраны (рис. 9.6) и вакуоли [Мосин И.М., 2000; Захарова Г.Ю., Зуева Т.В., 2001; Bengtsson В., Linder В., 1967; Miyake Y., Terasaki Н., 1999].Часто формируются гигантские кисты сетчатки (рис. 9.7), вдоль границ
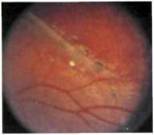
Рис. 9.4. Х-сцепленный врожденный (ювенильный) ретиношизис. Расслоенная сетчатка образует складку, идущую от лиска зрительного нерва в ннжнени сочном направлении. Атрофические пигментные нарушения и точечные очажки экссудата вдоль границы ретиношизиса в нижней половине сетчатки Локальные изменения сетчатки в виде очагов «битого металла».
которых откладываются пигмент и твердый экссудат (рис. 9.8). Эта форма заболевания, называемая буллезной, наблюдается приблизительно у 40 % детей раннего возраста с Х-сцеплен- ным врожденным ретиношизис ом и сочетается с косоглазием и нистагмом
[Мосин И.М. и др., 2001;
1948]. Приблизительно у 85 % детей с буллезной формой Х-сцепленного врожденного ретиношизиса кисты сетчатки самопроизвольно спадают (полностью или частично) через несколько месяцев или лет после момента их возникновения. На месте их первоначальной границы остаются демаркационные пигментные линии (рис. 9.9) [Мосин И.М., 2000; LevyJ., 1952; Basma- djian G. et al., 1973; Conway B.P., Welsh R.B., 1977; Greven CM. et al., 1980; Kawano K. et al., 1981; Verdagu- er J.T., 1982; Mosin I.M., 1998].
При биомикроскопии можно обнаружить, что ретинальные сосуды нередко как бы подвешены в стекловидном теле (рис. 9.10) или полостях между слоями расщепленной сетчатки, свободно проходя от слоя нервных волокон к наружным слоям сетчатки. В связи с этим витреоретинальные тракции часто приводят к механическим повреждениям сосудов, вызывая
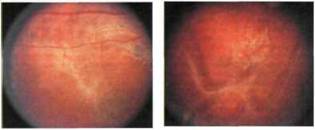
Рис. 9.6. и г - і,ѵ .к.-
вуали у ребенка £.Х-сцеплснным врожденным (ювенильным) ретиношизисом ь вуали прикрепляется артериола (в центре), свободно «подвешенная» и «плавающая» в стекловидном теле, вокруг локуса ее фиксации к сетчатке нанесены лазе ркоагуляты.
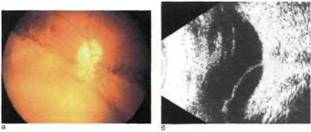
Рис.
9.7. Гигантская киста сетчатки у ребенка с Х-сцепленным врожденным (ювенильным) ретиношизисом в возрасте 1 года.л — офтальмоскопическая картина гигантская киста сетчатки с прозрачным содержимым в
нижней половине, пигментные демаркационные линии, располагающиеся над верхним краем кипы (в области ее первоначальной границы), б — эхограмма кипа сетчатки высотой 3,4 мм
массивные кровоизлияния в стекловидное тело (13—40 % случаев), ретро- витреальное пространство или в полости шизиса (20—29 %) [Мосин И.М. и др., 2001; Conway В.Р., Welsh R.B., 1977; Greven С.М. et ah, 1990; Ro- esch M.T. etah, 1998; Tasman W., 1999].
N.D.L. Со-.пр' и соавт. (описали 5 детей в возрасте до 2 лет с нистагмом и/или косоглазием и высоко проминирующим буллезным ретиношизисом, вовлекающим макулу Геморрагии в полость шизиса или стекловидное тело выявлены у 4 больных. При
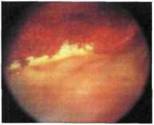
Рис. 9.8.
бенка с Х-сцепленным врожденным (ювенильным) ретиношизисом. Твердый желтый экссудат, располагающийся в полости ретин о шизиса и вдоль границы кисты, фрагмент демаркационной пигментной линии и атрофические трифокальные изменения в зоне проведения лазерной коагуляции.
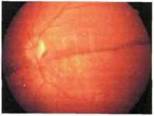
Рис. 9.9. Самопроизвольный коллапс гигантской кисты сетчатки у пебвнка с Х-сцепленным врожденным (ювенильным) ретиношизисом в возрасте 3 лет. Пигментная демаркационная линия вдоль границы самопроизвольно спавшейся гигантской кисты сетчатки

Рис. 9.11. Х-сцеплснный врожденный (ювенильный) ретиношизис. Аркообразный разрыв внутренних слоев сетчатки в области ретиношизиса, участки глиальной пролиферации, облитерированный сосуд.
Рис. 9.10. Васкулярные вуали мальчика с Х-сиепленным врожденным ретнноши- зисом в возрасте 2 лет. Вуали и нефиксированные артериолы и венулы в стекловидном теле, аркообразный разрыв внутренней стенки ретиношизиса н нижненаружном квадранте.
сроке м.;о І,і!.к'П!і;! от 5 до 6,5 лет во всех случаях отмечено полное прилегание буллезного ретиношизиса с формированием широкой пигментной демаркационной линии. При этом происходит незначительное повышение остроты зрения и уменьшение или исчезновение нистагма.
Автор длительно (от 3 до 11 лет) наблюдал 10 мальчиков с Х-сцепленным врожденным ретиношизисом, косоглазием и нистагмом, у которых в возрасте от 6 до 18 мес были выявлены макулярные изменения и одно- или двусторонние гигантские кисты сетчатки. За период наблюдения в 25 % глаз pin пились кровоизлияния в по- ь „ !і. ретиношизиса или стекловидное тело, которые полностью резорбировались в течение 3—8 мес. В 45 % глаз отмечен самопроизвольный коллапс гигантских кист с формированием в области их первоначальной границы широких демаркационных пигментных линий. В 35 % глаз регрессия кист произошла после лазерной коагуляции сетчатки. При осмотре детей в возрасте 6—12 лет установлено, что острота их зрения составляла 0,05—0,9 (в среднем 0,36 ± 0,23). Нистагм исчез у 4 больных. Ни у одного из 10 детей с буллезным ретиношизисом не было выявлено отслойки сетчатки к моменту завершения наблюдения [Мосин И.М. и
др., 2001].
При прогрессировании ретиношизиса в его внутренних слоях появляются множественные аркообразные разрывы (рис. 9.11), развиваются глиальная пролиферация и неоваскуляризация сетчатки, гемофтальм или кровоизлияния в полость кист, витреорети- нальные тракции приводят к формированию сквозных разрывов сетчатки.
Траклионная или регматогенная отслойка сетчатки развивается у 10— 16 % детей с Х-сцепленным врожденным ретиношизисом [Мосин И.М., 2000; Laatikainen L. et al., 1987; Ro- esch M.T. etal., 1998; Tasman W., 1999]. Описаны казуистические случаи развития экссудативной отслойки сетчатки [Regillo CD. et al., 1993; Fong D.S. etal., 1998].
Зрительные функции. Острота зрения у детей с Х-сцепленным врожденным ретиношизисом варьирует от светопроекции до 0,9. Н. Forsius и соавт. (1973) при обследовании 183 больных с Х-сцепленпым врожденным ретиношизисом определили, что средняя острота зрения у взрослых пациентов составляет 0,34. A.F. Deutman (1977) сообщил о 85-летнем пациенте с Х-сце-
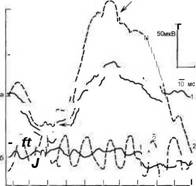
Рис. 9.12. Максимальный комбинированный ответ (а) и ритмическая ЭРГ (б) ребенка с Х-сцепленным врожденным ретиношизисом в возрасте 6 мес (1) и здорового ребенка того же возраста (2). Снижение амплитуды b-волны максимального комбинированного ответа и ритмической ЭРГ (стимуляция белой вспышкой с частотой 30 Гц) у больного.
По оси абсцисс — время анализа, мс; но оси ординат — амплитуда. мкН пленным врожденным ре- тиношизисом. острота зрения которого составляла 0,03 и 0,05.
М.Т. Roesch и соавт.
(1998) длительное время (в среднем 19,78 ± 8,74 лет) наблюдали за естественным течением заболевания у 92 пациентов с ретиношизисом и отмечали постепенное снижение средней остроты зрения с 0,3 при первом исследовании до 0,26 при заключительном осмотре. В 7 % случаев острота зрения у пациентов из этой группы уменьшилась до правильной светопроекции и ниже.
При периметрии выявляются полиморфные дефекты, соответствующие зонам ретиношизиса. Это могут быть квацрантопсия или концентрическое сужение. При статической периметрии порог яркостной чувствительности у большинства пациентов значительно повышен (на 0,6—1,0 log U), обнаруживаются относительные и абсолютные центральные, парацентральные и периферические дефекты [Мосин И.М., 2000; Захарова Г.Ю., Зуева М.В., 2001].
Диагностика. Диагноз устанавливают на основании данных офтальмоскопии. анализа семейного анамнеза и
. і-'К. Вспомогательные методы диагностики — ЭРГ, оптическая когерентная томография, ФАГ и эхоірафия.
При регистрации скотопической и фотопической ЭРГ отмечается выраженное снижение амплитуды Ь-волны, тогда как амплитуда а-волны сохраняется нормальной (рис. 9.12) |Шамши- нова Л.М., Волков В.В., 1998; Тапі- ло Т. et а!.. 1985; Peachey N.S. et a!., 1987; Shimazaki J., Matsuhashi M., 1987]. Снижение амплитуды b-волны ЭРГ свидетельствует о поражении внутренних слоев сетчатки, в частности клеток Мюллера) что коррелирует с данными морфологических исследований [Hirose Т. et а!., 19761. Преимущественное поражение клеток внутреннего ядерного слоя (биполярных и/или клеток Мюллера) также проявляется значительным снижением амплитуды волны N95 патгерн-ЭРГ [Papst N. et al., 1986; Clarke M.P. et al., 1996]. При прогрессировании ретиношизиса в патологический процесс вовлекаются фоторецепторы, что приводит к снижению амплитуды а-волны ЭРГ [Harris J.S., 1968; Vainio-Mattila BA. et al., 1969; Thaler A. et al., 1973; Forsi- us H. et al., 1973; Hirose T. et al., 1976].
У большинства пациентов с Х-сцеп- ленным врожденным ретиношизисом выявляют значительное снижение ам-
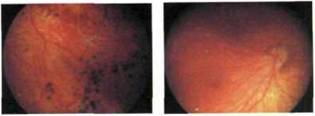
Рис. 9.14. Регрессивная ретинопатия недоношенных. Деформация диска зрительного нерва, гетеротопия диска, сосудистого пучка и макулы в нижневисочном иапраатснии
Рис. 9-13. Регрессивная ретинопатия недоношенных. Диффузные атрофические пигментные нарушения, участки пре- и интраретинального фиброза.
плитуды ритмической (см. [Г-н; Ті 2; и локальной ЭРГ, обусловленное кистозными изменениями в макуле [Мосин И.М. и др., 2001].
ЭОГ в начальной стадии заболевания, как правило, нормальная. Субнормальная ЭОГ встречается у пациентов в терминальной стадии болезни, сочетаясь со значительно редуцированной ЭРГ [DeutmanA-F., 1977].
При диагностике заболевания используют электрофизиологические методы, обладающие высокой чувствительностью. Нарушения ЭРГ в виде снижения амплитуды и/или задержки латентности b-волны фиксируют у 90—95 % носителей, не имеющих изменений на глазном дне [Berson E.L. et al., 1979; Gorin M.D., Arden G.B., 1991; HeckenlivelyJ.R., 1991].
При флюоресцентной ангиографии в начальной стадии процесса изменений в макуле нет [Саксонова Е.О. и др., 1979; Harris J.S., 1968; Vainio-Matti- 1а В.A et al., 1969; Deutman A.F., 1977]. По мере прогрессирования заболевания отмечают центральную гиперфлюоресценцию из-за дефектов пигментного эпителия сетчатки, а также повышенную проницаемость флюорес- цеина через стенки аномальных и новообразованных сосудов [Krause U. et al., 1970; Green J.L., Jampol L.M., 1979].
Дифференциальная диагностика.
В зависимости от тяжести нальных изменений необходимо дифференцировать Х-с це пленный врожденный ретиношизис от ретинопатии недоношенных, болезни Гольдманна— Фавра, болезни Вагнера, синдрома Стиплера, болезни Норри, семейной экссудативной витреоретинопатии, болезни Штаргардта, пигментного ретинита.
Ретинопатия недоношенных. Диагностика может вызывать затруднения при осмотре детей с регрессивной ретинопатией недоношенных III— IV стадий. У таких детей определяют диффузные пигментные нарушения, атрофические хориоретинальные очаги, области фиброзной пролиферации (рис. 9.13) и тракционную отслойку сетчатки. Диск зрительного нерва обычно деформирован, нейроретинальный край диска, сосудистый пучок и макула эктопированы в нижненаружном направлении (рис. 9.14), где на периферии выявляются зоны фиброза. ЭРГ у пациентов с ретинопатией недоношенных существенно снижена, иногда не регистрируется. Существенное значение для дифференциальной диагностики имеют результаты нейросоматического обследования и данные анамнеза (гестационный возраст детей при рождении меньше 29 нед, масса тела — меньше 1500 г).
Еще по теме Х-сцепленный врожденный (ювенильный) ретиношизис:
- Неполная ахроматопсия, или неполная врожденная сцепленная сХ-хромосомой колбо чковая дисфункция
- Анемия при стандартных методах терапии ювенильного ревматоидного артрита
- IХ.4.3. Х-сцепленное наследование
- 3.2. Клинико-лабораторные показатели у детей с ювенильным ревматоидным артритом
- Доминантный Х-сцепленный тип наследования заболевания
- 2.5.2. Прочность сцепления покрытия с металлической подножкой
- III.3.1. Сцепление генов
- 1.1.4. Возрастные особенности ревматических заболеваний у детей. Ювенильный ревматоидный артрит: клинико-патогенетические варианты
- Распространенность и тяжесть анемии у детей с ювенильным ревматоидным артритом
- Рецессивный Х-сцепленный тип наследования заболевания
- Х.1.3. Х-сцепленные рецессивные заболевания
- Врожденный кандидоз
- Моногенные болезни, имеющие сцепленный с полом рецессивный тип наследования
- IX.4.4. Y-сцепленное, или голандрическое, наследование