Фtнилпнровиногра дная олигофрения (фенилжетонурия)
Фенилпнровиноградная олигофрения представляет собою врожденное, генетически обусловленное нарушение метаболизма фенилаланина, вызывающее в конечном итоге тяжелое нервно-психическое заболевание.
Впервые это заболевание описал в 1934 г. Foiling в Норвегии. В дальнейшем заболевание наблюдалось во многих странах. По данным статистических исследований, произведенных в разных странах, частота фе- нилкетоиурии колеблется от 2 до 6 больных на IOOOOO населения. Это заболевание распределяется неравномерно. Так, оно чаще встречается в Норвегии и реже во Франции. Среди новорожденных фенилкетонурия встречается у 1 на 40 000 родившихся. Лина мужского н женского пола болеют одинаково часто, Среди больных психиатрических учреждений количество паниен-
тов с пировиноградной олигофренией колеблется от 0,5 до I %.
Клиническая картина. Психомоторное развитие детей в течение первых месяцев жизни кажется нормальным, но затем оно начинает отставать. Больные начинают самостоятельно сидеть—к I—2 годам. Одна треть детей не научается ходить, а остальные начинают ходить после 2 лет. Половина больных вовсе не владеют речью, другие повторяют только слышимые слова (эхолалня). У говорящих детей развитие речи задержано и она появляется между тремя и четырьмя годами жизни. Иногда у больных отмечается отставание роста, веса и окружности головы. Задержка умственного развития вследствие феннлпировииоградной олигофрении может быть выражена в различной степени от дебильности и имбецильности до идиотин. Нормальное умственное развитие у нелеченньтх больных встречается в виде исключения. Интеллектуальная отсталость редко сопровождается чрезмерной двигательной активностью или агрессивным поведением.
Характерен внешний вид больных. У 80% детей кожа мало пигментирована, нежная, сухая, полосы светлые, глаза голубые. Сравнительно часто наблюдаются различные сыпи.
У детей раннего возраста кожные поражения встречаются в 20%. Больные обычно сидят, скрестив ноги, при ходьбе голова и туловище несколько наклонены вперед, ноги широко расставлены и слегка согнуты в коленных и тазобедренных суставах. У 2/з больных обнаруживается ригидность и мышечная гипертония экстрапирамидиого типа. Сухожильные рефлексы живые. В виде исключения наблюдаются спастические параплегии и положительный симптом Бабинского. Эпилептические припадки бывают у 1Λ — *∕3 больных. Затем эти припадки у старших детей становятся реже.У больных фенилкетонурией часто отмечается «мышиный запах», зависящий от выделения с мочой фенилуксусной кислоты.
Лабораторные исследования. Наиболее простой является реакция с хлорным железом.
К. 5 жл свежей мочи добавляют несколько капель 10% FeClj. Предварительно моча должна быть приведена к pH — 6. Через несколько минут появляется зеленое окрашивание, которое 5
і и IIIiJi і іініібч.'іі.іінй Iiiiil-Iichiiiiiiiiii ∣ic∣∣c i .,0 мчи Лоложнтель- IlJll ,.Ii JMlllJ y∣.J.Il,IllilCT Illl ll∣llll,y ι,c tunc n Ml)lll, ι∣∣c іііі.ішіроиино- l∣ιj,,lllllιi КИСЛОТІ,I.
Применяется также проба, известная под названием PheflistlX,
Фильтровальная бумага, пропитанная особым раствором (ферроаммонпецый сульфат, сульфат магния п циклогексосуль- фопиая кислота), опускается в исследуемую мочу; при положительной реакции появляется не позже НО с сине-зеленая окраска.
C целью массового обследования новорожденных применяется и микробиологический тест Гатри, определяющий уровень фенилаланина в крови {Б. В. Лебедев и М. Г. Блюмина).
Для этого заболевания характерно накопление и органических жидкостях фенилаланина н его дериватов. В суточной моче количество /-фенилаланина достигает 1 г, в то время как в норме он выделяется в количестве меньшем чем ЗО ліз.
Кроме феннлпиро- виноградпон кислоты, с мочой выделяется фенилмо- лочная и фенилуксусная кислота и ацетнлфенилглута- мип.Моча содержит еще дериваты О-тирозина: О-гид- роокспфенмлппруват, лактат, ацетат и дериваты триптофана, индоллактат и индолацетат; последние выделяются ежедневно в количестве 0,02—0,15 мг, Количество фенилаланина в крови больных превышает в 30 раз содержание его у здоровых, норма для которых равна 0,17—1,5 мг% (Jervis), За исключением фенил- ннровипоградной кислоты, другие метаболиты фенилаланина, выделяемые в избытке с мочой, находятся в кроки в значительно меньшем количестве, что связано с быстрым выведением их почками. Иногда при электрофорезе можно обнаружить и изменения β-липопро- теинов. В спинномозговой жидкости больных имеется повышенное количество фенилаланина — до 8 ,мг%, вместо 1 мг% в норме. При датологоанатомических исследованиях морфологических изменений в нейтральной нервной системе, характерных для этого заболевания, обнаружить не удалось. У части больных, однако, отмечается более или менее выраженная демиелинизация, недоразвитие дендритов и увеличение гл и оз а мозга.
Патогенез. Фенилаланин является составной частью почти всех белков. Обычно у здоровых детей он переходит в тирозин, вследствие окисления, HO при φe∏H.1- кетонурнн фенилаланин подвергается дезаминированию и переходит в пировиноградную кислоту И Другие дериваты, которые выделяются с мочой. Метаболизм у больных схематически может быть представлен следующим образом:
фенилаланин → феиилпиропиноградная кислота → φenn.τ⅛.104. пая кислота → фенилацетилглютамин.
При фенилкетонурии содержание фенилаланина в различных белках организма остается нормальным, за исключением некоторых белков сыворотки крови. У больных блокирован переход фенилаланина в p.γ∣1- розип. Эта блокада вызывает накопление фенилаланина в жидкостях организма. Количество р-тнроэццз не увеличивается после дачи с пищей / фенилаланина, как это наблюдается у здоровых людей.
Однако, если больным назначается меченый фенилаланин, некоторое количество меченого тирозина можно обнаружить н крови, равное, примерно, 10% количества, получаемого у здоровых. Эти исследования показывают, что при фенилкетонурии имеется неполная блокада фенилаланина. Jervis показал, что кусочек печени, взятый у больного, не в состоянии окислить фенилаланин, в то время как инкубация при тех же условиях с печенью здоровых людей вызывает его окисление. Система гидроксилирования состоит из двух фракций: I фракция, лабильная, находится только в печени и ∏ фракция, стабильная, находится в других органах (мозг, ночки). При кетонурии инактивирована или отсутствует I фракция гидроксилирования.В настоящее время установлено, что блокада фенилаланина обусловлена ферментативной недостаточностью феиилпарааминогидроксилазы. В патогенезе болезни первичным является ферментативная блокада гидроксилазы; в результате этого фенилаланин накапливается в организме больного и выделяется в повышенном количестве мочевыводищей системой. Одновременно с увеличением в крови фенилаланина наблюдается некоторое снижение уровня тирозина. Дальней- шее выравнивание количества тирозина может
происходить насчет нищи больною, если она содержит достаκ∣ι∣noc количество этой аминокислоты. Кроме первичной блокады гидроксилазы, при фенилкетону- ріін отмечается также инактивация тирозиназы, приводящая к снижению уровня адреналина, норадреналина и меланина. Появление в большом количестве фенилаланина и его дериватов в моче объясняется тем, что количество фильтрующегося фенилаланина и фе- нилпировиноградной кислоты намного превышает способность обратной реабсорбции их с мочой дериватов о-тнрозина и соединений индола. Предполагается, что окисление фенилаланина, которое в норме ведет к образованию р-тирозина, может в части случаев привести к образованию о-тирозииа. Это видоизменение реакции (фенилаланин →-о-тирозин), вероятно, происходит под влиянием определенного специфического фермента, пока еще не выделенного.
При нормальных условиях метаболизма эта реакция имеет весьма ограниченное значение. Однако можно предполагать, что когда обычные пути обмена фенилаланина блокированы, часть его трансформируется по добавочным путям, образуя о-тирозин и его дериваты. В последнее время при фенилкетонурии уделяется много внимания вторичным нарушениям метаболизма триптофана. Как уже указывалось, при этом заболевании легко возникают процессы дезаминирования. Если дезаминированию, подвергается триптофан, то последовательно образуются индолпировикоградная, индолуксусная и ин- долмолочная кислоты. Образование этих дериватов ведет к избыточному выделению их с мочой у больных фенилкетонурией. Интересно отметить, что эти индо- ловые соединения исчезают при назначении больным диеты, бедной фенилаланином. Дальнейшие исследования выяснили, что, кроме того, имеется нарушение перехода триптофана в серотонин. В норме этот переход осуществляется следующим образом: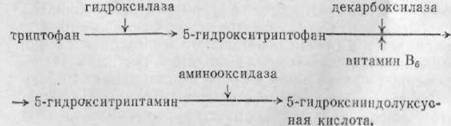
Последняя выводится с мочой (Alozziconacci с COiiBT.). У больных феннлкетонурией установлено снижение серотонина и уменьшено выделение C мочой количества 5-гидроксннндолуксусной кислоты. Исследования Mozziconacci л др. показывают, что психоневрологические расстройства при фенилкетопурин непосредственно связаны с нарушением метаболизма триптофана, вызванного блокадой перехода фенилаланина в тирозин. Взаимоотношение между нарушением метаболизма аминокислот (фенилаланина, тирозина и триптофана) и клиническими симптомами еще недостаточно выяснены. Известно только, что при питании пищей, бедной фенилаланином, у больных улучшается неврологическое состояние и уменьшаются поражения кожи. По-видимому, клинические симптомы заболевания зависят от биохимических изменений метаболизма.
Между степенью снижения интеллекта и уровнем в крови фенилаланина и его дериватов зависимости не обнаружено. Отсутствует также корреляция между степенью умственной отсталости и количеством дериватов фенилаланина, выделенных с мочой.
Кроме того, не выяснено, имеется ли какая-нибудь определенная зависимость между степенью умственной отсталости и выделением с мочой фенилацетоглютамина или ин- доловых соединений, а, между тем, клиническое значение последних заслуживает внимания, так как при других заболеваниях, сопровождающихся кожными, нервными и психическими поражениями (например. при пеллагре) выделение индоловых соединений тоже увеличивается. Некоторые клинические симптомы могут быть объяснены недостатком в организме тирозина, вызванным блокадой фенилаланина. Слабая пигментация волосу больных связана с недостаточным количеством тирозина, из которого образуется пигмент меланин. При увеличении в пище тирозина волосы у больных могут потемнеть. Возможно, что при фенил- кетонурии имеется также понижение активности тиро- знназы, которая способствует образованию меланина из тирозина. К, Ланг и др. объясняют поражение центральной нервной системы недостатком тирозина.При фенилпировиноградНбй олигофрении одинаково нарушен метаболизм L- и Р-фенилаланнна. Среди
•I· I l∣'lt∣.ΗIInh фсІІІІ IfMhlIIIIIiI .......................... .... ....... II1IIia фенил-
M-M1CIHHI Ml· ΙΟΙ.I, ()6yc,IfllljllIhiHIIIIMtI HI,Illlllllhlfl запах Мичи больных. Оди,'mo rto,∣ι∣,IIifHi часи, фенилаланина, .,ll' IIMIIIIhpy IoillhIlCII 11 печени, IipehpIIlHfH-H H Il !піровиноградную кислоту, которая выделяется мочой.
PeiKIMIIpyil, следует еще раз подчеркнуть, что в патогенезе феиилкетонурни основным является ферментативная блокада фенилаланина, вызывающая ряд вторичных биохимических нарушений обмена тирозина и триптофана. Нарушения метаболизма последнего вызывают снижение синтеза серотонина, играющего существенную роль в нормальной жизнедеятельности ЦНС.
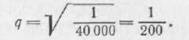
Наследственность. Известно, что всякий энзим связан с определенным геном. При мутации гена энзим изменяется в своей активности или лишается специфического действия. Фенилкетонурия является одним из примеров такой зависимости между энзимом и геном. Как показали исследования Penrose и других, эго заболевание генотипически рецессивное. Больные рождаются у здоровых родителей, которые являются гетерозиготными по данному гену и часто находятся в кровном родстве. Количество родственных браков при феиилкетонурни оказалось увеличенным до 6—10%; как уже указывалось, частота заболевания фенилкето- нурией равна 1 на 40 000 родившихся. Исходя из этого, можно вычислить частоту гена:
Частота для гетерозиготов это значит, что
это значит, что
на IOO человек населения имеется один, являющийся носителем латентного гена.
Интересно, что у женщин, больных фенилкетону- риен, могут рождаться здоровые дети.
Лечение. Единственной эффективной терапией фс· нилкетонурии является питание пищей, бедной фенилаланином. Назначение больным тирозина, без ограничения фенилаланина, не оказывало общего благоприятного действия, хотя цвет волос при длительном лечении тирозином темнел. Назначение питания со значительным ограничением фенилаланина является
IU
нелегкой задачей, так как эта аминокислота входит почти во все белки. Для приготовления диеты с ограничением фенилаланина применяются гидролизаты белков. Полное исключение фенилаланина из пищи оказалось вредным для больных детей. Оптимальные дозы фенилаланина: 25 мг на 1 кг веса для больных грудных детей и 5—15 мг— для детей старшего возраста (в сутки). При такой диете необходимо часто проверять количество фенилаланина в крови с целью сохранения его нормального уровня. Наряду с питанием гидролизатами белков необходимо назначать больным достаточное количество жира, углеводов (сахара), минеральных солей, микроэлементов и витаминов. Для грудных детей рекомендуется следующая диета: IOOO г гидролизата казеина, в котором резко снижена концентрация фенилаланина. Назначается гидролизат (кетонил), который содержит /-тирозина — 17 г, (//-триптофана — 7,2 г, /-цистина— 12,5 г, а также (//-метионин, /-гистидин, глицин и холин. Эта смесь разбавляется горячей водой и в нее добавляют соли и жир. Суточная доза делится на три приема и хранится в сухом помещении в закрытых сосудах.
Применяется также лечение отечественным препаратом— гипофенатом (Б. В. Лебедев и М. Г. Блюмина).
При приготовлении пищи для детей старшего возраста рекомендуются следующие продукты: мед, сахар, масло, печень (3 раза в неделю по 20 г), абрикосы, яблоки, груши, виноград, арбузы, салат, лук, огурцы. Запрещаются мясо, молоко, сыр, рыба, бобовые, яйца, картофель, финики, изюм, шоколад. Животные и растительные белки содержат' от 4 до 6% фенилаланина. Так, например, 100 г молока содержит 150—170 мг фенилаланина; 100 г сырого мяса и печени содержит от 650 до 1200 мг фенилаланина, а 100 г гидролизата казенна содержит фенилаланина от 10 мг (цимогран) до 100 мг (кетонил). Терапевтический эффект диеты с ограничением фенилаланина весьма благоприятен: через несколько дней у больных исчезает из мочи фенилаланин и фенилпировнноградная кислота. В сыворотке крови фенилаланин снижается до нормы в течение 3—6 недель (Mazziconacci с соавт.). Отрицательными чертами диеты, содержащей гидролизаты,
n∣' IiH iCM ее однообразие и неприятный вкус. Под и HIhhhcm такой диеты у детей может появиться рвота и понижение аппетита. Наблюдения Bickel и др. показали, что умственное развитие детей, леченных дне- юй с ограничением фенилаланина, нормально или почти нормально, если терапия начата в первом полугодии жизни. Более позднее лечение обыкновенно оказывает менее значительный эффект. Если лечение начато поздно, после 5 лет, отсталость умственного развития у больных не ликвидируется.
Для иллюстрации приводим краткие данные о девочке 4 лет, лечившейся в нервном отделении ЛПМИ.
Больная С. В., 4 лет, поступила в клинику 14 октября 1968 г. по поводу резкого отставания в психическом развитии. Девочка родилась в срок, закричала сразу и взяла грудь при первом прикладывании. Период новорожденное™ протекал нормально. Мать считает ребенка больным с 7—8 месяцев, когда появились судорожные припадки В это же время замечено отставание психомоторного развития. Сидит с 8 месяцев. Самостоятельно стала ходить с 2‘/г лет. Первые слова произносит с З'/г лет. Припадки носят тонический характер. Последнее время они участились до 5—6 раз в день.
Родители здоровы, работают а совхозе на полевых работах. Кровное родство отрицают. Отцу 28 лет, матери 34 года. В семье имеется еще один мальчик 5,5 лет, который страдает аналогичным заболеванием. При осмотре у девочки отмечаются светлый цвет кожи, очень светлые волосы и голубая окраска радужных оболочек глаз. Внутренние органы и пределах нормы. В психическом развитии обнаружено резкое отставание. Девочка не знает матери, заданий не выполняет, самостоятельно не ест, неопрятна, у нее отмечаются однообразные автоматические движения. Активной речи нет, произносит отдельные слова. Склонна к агрессивным действиям. При исследовании нервной системы патологические рефлексы не обнаружены. Парезов и параличей нет. Мышечный тонус не изменен.
Обычные клинические исследования крови, мочи и кала нормальны Реакция мочи с хлорным железом резко положительна, вследствие наличия в моче фепилпировиноградпой кислоты.
На основании клинической картины и положительной реакции с хлорным железом, а также аутосомной рецессивной наследственности, у девочки диагностирована феннлпировикоград- нап олигофрения
Альиантопурии
Это заболевание встречается редко. Частота его колеблется между 1 X КН6 и 1 ? IO-7 (Hogben с сотр.). По данным В. П. Эфроимсона, эта аномалия имеет частоту порядка 3—5 X IO^Λ
Алькаптонурия стала известна раньше других наследственных эизимопатий. Свойство мочи некоторых людей темнеть при соприкосновении с воздухом было известно уже в XVi в. Scribonius первый описал аль- KaiiTOHypniO в 1584 г. Baedecker в 1859 г. высказал предположение, что свойство мочи темнеть связано с наличием в ней специфических веществ неизвестной природы. Эти вещества получили название алькал- тона, а аномалия выделения темнеющей мочи была названа алькаптонурией. Волков и Бауман в 1891 г, показали, что алькаптон представляет собою гомогеи- т из и новую кислоту, Qarrod в 1902 г, доказал наследственный рецессивный характер этого заболевания. Алькаптонурия чаще встречается у мальчиков, которые болеют в 2 раза чаще, чем девочки. В литературе описано свыше 300 заболеваний алькаптонурией.
Клиническая картина. Заболевание проявляется сразу после рождения ребенка, однако диагноз аль- KaiiTouypiiH в период новорожденное™ устанавливается редко. По литературным данным, Заболевание у новорожденных описано только у 12 детей.
В грудном возрасте основным симптомом болезни является изменение цвета мочи на пеленках. Вначале после мочеиспускания пеленка сохраняет нормальный желтый цвет, затем происходит изменение окраски на красный, который появляется тем быстрее, чем выше концентрация гомогентизиновой кислоты в моче. Интенсивность красной окраски различна и более выражена по периферии пятна. Через несколько часов вокруг пего на пеленке появляется темноватая кайма.
Алькаптонурия чаще распознается у старших Детей. Заболевание сохраняется всю жизнь и сущность его заключается в потемнении мочи вскоре после ее выделения. Общее состояние больных ие нарушается. Единственным осложнением является охроиоз, при котором происходит отложение темпо-желтого пигмента в некоторых тканях вследствие накопления в них продуктов окисления гомогентизиновой кислоты. Эта пигментация выражена главным образом в хрящах ушей, носа и суставов. На склерах, роговице и конъюнктиве глаз появляются синеватые пятна. Значительно реже поражаются десны, кожа и ногти. Охроноз, как
осложнение алькаптонурии, появляется у больных в более позднем возрасте.
Больные охронозом большей частью чувствуют себя хорошо. Однако при значительных отложениях пигмента в суставных хрящах вторично наступают дегенеративные изменения суставных поверхностей и межпозвонковых дисков н развивается охроноэный артроз. Артрозы при алькаптонурии чаще наблюдаются у мужчин и обычно сопровождаются ригидностью позвоночника. Сильные боли при этом отмечаются только у половины больных. Наиболее часто охронозом поражаются плечевые суставы, реже коленные и тазобедренные. Мелкие суставы изменяются редко. На рентгенограммах позвоночника определяется уплощение межпозвонковых дисков и отложение извести в поясничных н грудных отделах. Позвоночные тела не поражаются, но иногда наблюдаются остеофиты. Перя- артикулярные кальцинаты могут быть обнаружены также вблизи других суставов. Кроме пигментации хрящей, у больных часто отмечается темная ушная сера; эта аномалия относится к ранним проявлениям алькаптонурии и наблюдается у детей. Пот в подмышечных и паховых областях тоже может быть окрашен в черный цвет.
Лабораторные исследования. Моча больных, оставленная на воздухе, через некоторое время темнеет. Моча после подщелачивания восстанавливает реактив Фелинга даже на холоде; вначале образуется кирпично-красная окраска, которая затем темнеет и переходит в коричнево-зеленоватую. При добавлении к моче нашатыря и поташа быстро наступает стойкое потемнение. Если к моче добавляется азотнокислое серебро и нашатырь, происходит восстановление металлического серебра. Добавление полуторахлористого железа изменяет желтый цвет мочи на зеленый. C реактивом Ми л лона образуется лимонно-желтый осадок, чернеющий при нагревании. Алькаптонурическая моча разлагает светочувствительный слой фотографической бумаги. В темноте пробирка с мочой люминесцирует. Количественное определение гомогентизнновой кислоты в моче проводится по методу Метца. Количество этой кислоты в моче больных алькаптонурией колеблется от 0,1 до 2%. Уровень гомогентизнновой кислоты на
ходится в зависимости от количества белков в пище; при вегетарианском режиме он может значительно снизиться.
Существует определенная корреляция между выделенным количеством гомогентизнковои кислоты и количеством общего азота, поэтому отношение гомоген- тизиновой кислоты к общему азоту является удобным показателем для характеристики нарушения белкового обмена. Этот показатель приблизительно один и тог же у всех больных алькаптонурией при одинаковой пище. Количество гомогентнзнновой кислоты возрастает при увеличении в пище фенилаланина и тирозина, а также при повышении температуры тела у больных и лечении их глюкокортикостероидами. Кроме ненормального выделения гомогентизиновой кислоты с мочой, других нарушений белкового обмена не наблюдается. Концентрация гомогентизиновой кислоты в крови при алькаптоиурии окончательно не выяснена. Предполагается, что ее концентрация должна быть около 3 мг на I л.
У больных алькаптонурией иногда отмечается положительная реакция Вассермана, несмотря на отсутствие заболевания сифилисом.
Патогенез алькаптоиурии обусловлен нарушением обмена в организме фенилаланина и тирозина. Гомо- гентизнновая кислота (диоксифенилуксусная кислота) является нормальным метаболитом обмена тирозина.
Работами с меченым углеродом (Grecnherg) доказано, что вышеуказанные изменения фенилаланина происходят в печени, в которой благодаря воздействию специфических ферментов большую роль играют реакции переаминирования, декарбоксилирования и замены гидроксильной группы в боковой цепи.
Недавно из гомогената крысиной печени было выделено две энзимные фракции; одна из них переводила гомогентизнновую кислоту в фумарилацетоуксусную, другая фумарилацетоуксусную кислоту в фумаровую и ацетоуксусную кислоты. Suha и Taked[I] выделили из печени кролика энзим гомогентизиназу, которая переводила гомогентизнновую кислоту в фумарилацетоуксусную кислоту.
I Ipll IljlbKilllTOHypiin не происходит конечных ИЗМЄ· HrHllf1I 1'ОМОГЄНТИЗИНОБОЙ кислоты и потому этот метаболит выделяется с мочой в неизмененном виде. У здоровых людей прием внутрь гомогентизиновой кислоты не сопровождается выделением ее с мочой, так как в организме происходят метаболические изменения этой кислоты. Дигидроксифенилаланин значительно увеличивает выделение гомогентизиновой кислоты.
Количество гомогентизиновой кислоты в крови не увеличивается при алькаптонурии и не превышает 3 λic‰; клиренс гомогентизиновой кислоты у больных повышен и равен 400—500 мл в I мин. Предполагается, что гомогентизиновая кислота не только фильтруется через клубочки почек, но и секретируется в канальцах. Neuberger с сотр, и Martin с сотр. считают, что при алькаптонурии имеется тубулярная аномалия почек. При дифференциальной диагностике следует иметь в виду меланурию, которая наблюдается при злокачественных опухолях.
В отличие от врожденной лорфирни при алькаптонурии отсутствуют кожные изменения. От наследственной алькаптонурии следует отличать симптоматические формы, при которых увеличивается выделение гомогентизиновой кислоты с мочой вследствие недостатка витамина С, как это наблюдается у недоношенных детей в первые недели жизни.
Наследственность. Алькаптопурия представляет собою наследственное расстройство метаболизма, вызванного недостаточностью гомогентизиназы. Обычно передача происходит рецессивно (Hogben, В. П. Эф- ронмсон). Кровное родство родителей обнаружено в 18%. Однако в литературе имеется описание семей н с доминантной наследственностью (Milch и Milch). Для объяснения характера наследственности при аль- каптонурии большинство авторов придерживаются гипотезы, что генетически существуют два варианта болезни, различить которые клинически не представляется возможным, Klein считает, что наследственность генетически связана с полом. Большинство авторов отрицает такую связь. Преобладание среди больных мальчиков (a∕3) доказано статистически.
Лечение. У большинства больных можно ограничиться симптоматическим лечением. Для лучшего
окисления в организме тирозина полезно назначение аскорбиновой кислоты. Эффект от такого лечения указывает на нарушение метаболизма / тирозина (Л. Ранен и coτp.). В качестве иллюстрации приводим собственное наблюдение.
Больной М. А., 9 лет, поступил в клиническое детское отделение Областной больницы 30/1 1964 г. Жалобы на выделение мочи, которая затем темнеет. Родители здоровы. В семье трое сыновей, нз них один здоров, а двое других (9 и 3 лет) выделяют мочу, темнеющую па воздухе н приобретающую темно- коричневую окраску. Эта особенность мочи наблюдается с рождения. Моча на пеленках приобретала коричневый оттенок и плохо отстирывались. Среди родственников семьи такая аномалия неизвестна. При клиническом обследовании со стороны внутренних органов отклонений от нормы не обнаружено. Физическое и умственное развитие мальчика соответствует возрасту. Морфологическое исследование крови без особенностей. Холестерин — 150 лг%, RW — отрицательна. Обычное исследование мочи в пределах нормы. При дополнительном исследовании мочи получены следующие результаты: 1) на воздухе моча становится темной, буровато-черного цвета; 2) после добавления децинор- мального раствора NaOH моча сразу приобретала темно-коричневый цвет; 3) после добавления к моче реактива Фелиига наступало восстановление реактива как па холоду, так и при подогревании; 4) после добавления 2% раствора AgNO3 появлялся зеленоватый оттенок мочи, а добавление нескольких капель нашатырного спирта быстро приводило к восстановлению металлического серебра.
При количественном определении гомогеннзнновой кислоты по способу Метца у больного оказалось в моче 1,2% гомогенти- зионовой кислоты.
Больному вводилось ежедневно внутривенно 500 мг аскорбиновой кислоты, после чего реакция мочи с NaOH и реактивом Фелиига стала отрицательной, а с AgNO3 — слабо положительной.
Альбинизм
(ахромия, лейкопатия)
Это заболевание известно с древних времен и впервые было описано римскими авторами (Плинием и Помпеем). Заболевание относится к редким. Частота его равна в европейских странах 1 : IOOOO—I : 20 000 населения. Альбинизм описан среди всех пародоп и во всех странах мира; он встречается не только среди людей, но также в животном и растительном мире. Клинически различают универсальную иди полную ахромию и частичную црти ограниченную φω⅛ui⅛ когда
отсутствие пигмента наблюдается только на некоторых участках тела.
Клиническое понятие о полном альбинизме не находит подтверждения в современных исследованиях. При электронной микроскопии кожи, альбиносов можно обнаружить частично меланнзированлые мелано- сомы. Имеются и небольшие количества тпрозиназы.
Таким образом, правильнее говорить не о полном отсутствии тпрозиназы, а о значительном уменьшении ее активности. Клиническим подтверждением этих исследований является различный по интенсивности цвет глаз и волос у больных «полным» альбинизмом. Однако термин полного альбинизма сохраняется в клинике с целью противопоставления частичному альбинизму, который тоже встречается изредка (1 : 25 000 населения).
Обычно в более раннем возрасте при альбинизме кожа и внутренние органы почти лишены пигмента, а в период полового созревания полосы и глаза становятся чуть темнее. Наследственность при полном альбинизме аутосомная — рецессивная. Мужчины и женщины болеют одинаково часто. При частичном альбинизме наследственность — доминантная.
Этиология и патогенез. Альбинизм развивается вследствие врожденной недостаточности активности энзима тпрозиназы в меланоцитах. Меланоциты— это специализированные секреторные клетки, от которых зависит в основном окраска кожи человека. Располагаются они в базальном слое эпидермиса. В человеческом организме около 2 биллионов меланоцитов. В патогенезе альбинизма имеют значение число меланоцитов и распределение их в организме, а также механизм и контроль за синтезом меланина, который является секретом меланоцитов и образуется при воздействии энзима тпрозиназы.
Меланин защищает кожу от патологического воздействия солнечных лучей. Кожа повреждается при облучении лучами ультрафиолетового спектра. Непосредственный эффект такого облучения выражается в кожных изменениях от эритемы до ожоговых пузырей. При продолжающемся воздействии ультрафиолетовых лучен в течение длительного времени наступают хронические изменения кожи в виде сухости, потери эла-
сгичноети, атрофии, телеаигеэктазин, морщинистости. Механизм этих повреждений пока неизвестен. Предполагается, что в основе лежит изменение секрета, вырабатываемого мелаиоцитами. Мелании предохраняет кожу от вредного влияния солнечных лучен. Вероятно, он нейтрализует патологический секрет, выделяемый при радиации: усиленный меланогенез возникает при длине волны ультрафиолетового спектра — 290— 320 JH-MK-M.
Однако обычный меланогенез может происходить и под влиянием видимого света с длиной волны свыше ООО ммкм. При образовании меланина вначале выделяется светло окрашенный секрет, который в дальнейшем, благодаря окислению и другим изменениям, превращается в темный пигмент. Предполагается, что это происходит под воздействием спектра с длиной волны от 300 до 700 ммкм. Для окраски кожи имеет значение не столько количество меланоцитов на данном участке кожи, сколько их способность продуцировать меланин.
Тпрозиназа является энзимом, содержащим медь, который катализирует окисление тирозина в дегидрооксифенилаланин (Допа) и дальнейшее окисление в допахинон. Клетки меланоцитов, которые под влиянием тирозиназы могут перевести Допа в нерастворимый темный пигмент, носят название Допа-положн- тельных клеток. Дальнейшие изменения сводятся к образованию тирозин-меланина. Этот пигмент легко соединяется с белками. Резюмируя можно сказать, что альбинизм является следствием наследственной энзн- мопатни, при которой отсутствуют меланоциты, способные синтезировать меланин-тирозиназу. Однако у альбиносов никаких морфологических аномалий в ме- ланоиитах не обнаруживается даже при электронной микроскопии. Защитная роль меланина отсутствует при альбинизме, вследствие чего у больных наблюдается повышенная чувствительность кожи к солнечным лучам и понижается зрение.
Клиническая картина. Основные симптомы альбинизма у людей зависят от почти полного отсутствия меланина в различных органах. Кожа больных отличается нежностью и бледно-розовым оттенком, зависящим от просвечивания капилляров. Придатки кожи также имеют характерные особенности. Волосы на го
лове, брови, ресницы И волосы на теле светло-белокурые. Ногти нередко ломкие. Глаза у альбиносов имеют целый ряд изменений. Зрачки у них с красноватым блеском. Радужная оболочка светло-голубая. Сосудистая оболочка и сетчатка лишены пигмента. Глазное дно оранжевого цвета и хорошо заметны хореоидаль- ные сосуды. Всегда имеется горизонтальный нистагм, сопровождающийся понижением зрения, вследствие близорукости. Иногда имеются микрофтальмия, отсутствие хрусталика или fovea centralis.
У альбиносов имеется фотофобия, выраженная в различной степени, поэтому они ходят опустив голову с полузакрытыми глазами. Глаза у них быстро устают, может наступить амблиопия (Рачсв с соавт.). Нередко у них наблюдаются конъюнктивиты. Обычно умственное развитие у альбиносов не страдает, но в связи с понижением зрения, оно может быть замедленным.
По данным некоторых авторов, альбиносы обычно ниже ростом, чем их сверстники. При альбинизме могут наблюдаться и другие врожденные аномалии — полидактилия, глухота, эктромелия и др.
Из лабораторных исследований отмечается поздняя лероксидазная реакция в циркулирующих лейкоцитах.
При дифференциальном диагнозе следует исключить пировиноградную олигофрению. Вторичные витилиго и лейкодерму отличить от частичного альбинизма легко, так как они являются приобретенными последствиями сифилиса, трихофитии и других заболеваний, а частичный альбинизм — врожденная аномалия, проявляющаяся уже в период новорождепности. Лечение альбинизма неизвестно.
Приводим краткую выписку нз истории болезни больного, которого мы наблюдали в детской Бассейновой больнице г. Ленинграда.
Больной Б. Г., 15 лет, поступил в хирургическое отделение 18 января 1972 г. по поводу альбинизма, горизонтального нистагма и врожденного укорочения (эктромелия) правого бедра.
Болен с рождения. C 11 лет учится в специлизированной школе-интернате для слабовидящих детей.
Ilpn осхютре отмечается своеобразная походка вследствие укорочения правой нижней конечности. При ходьбе опирается на пальцы правой ноги. Правая коленная чашечка ротирована кнаружи. Правое бедро с рождения укорочено и утолщено. Длина правого бедра— 29 с.и, левого бедра— 43 см. Мышцы правой
голени атрофичны. Осмотрен ортопедом. Остановлена врожденная эктромелня правого бедра. Внутренние органы Qea особенностей, Горизонтальный нистагм и понижение зрения. Диски зрительных нервов бледноваты, контуры нечетки. Артериальные си- еуды несколько сужены, вены слегка расширены. Ски о с коші чески: миопия средней степени. Острота зрения—0,1. Стекло не улучшают зрения. Антропометрические данные: рост 162 см, рост сидя 81 см, окружность грудной клетки 92 см, окружность головы 55 с.«, вег 54,3 кг. Исследование крови при поступления: эр. 4 700 000, Hb 82 ед., л. 5000, э. 3, п. 5, с. 65, лимф 23, мои. 4, РОЭ 7 мм а 1 ч. Коагулограмма без особенностей. Моча нормальна. Радужная оболочка голубого цвета с небольшим количеством пигмента.
Заключение: альбинизм, горизонтальный нистагм, частичная атрофия зрительных нервов. Миопия средней степени. В дальнейшем больному произведена операция остеотомии с целью удлинения правого бедра.
липопдозы
К заболеваниям с нарушениями липидного обмена, или к липоидозам, относятся редко встречающиеся болезни, при которых имеются отложения специфических липоидов в ретикулогистиоцитарной системе. К этим заболеваниям относятся 4 болезни: 1) болезнь Гоше, 2) болезнь Пик-Нимана, 3) болезнь Тей — Сакса и 4) гиперхолестерииемическнн ксантоматоз. При болезни Гоше имеются отложения липоида-керазииа во внутренних органах. При болезни Пик — Нимана в тех же органах откладывается сфингомиелин, а в мозгу ганглиозид. При болезни Тей — Сакса происходит отложение ганглнознда в мозгу и сетчатке глаза (в ганглиозных клетках и нейроглии). При гиперхолесте- рннемическом ксантоматозе в коже, сухожилиях и кровеносных сосудах откладывается холестерин и лецитин. Весьма вероятно, что при сфингомиелинлипоидозах (болезни Гоше, Пик—Нимана и Тей —Сакса) имеется первичное генетическое расстройство обмена веществ.
Предполагается, что при этих заболеваниях нарушается расщепление липоидов; однако не исключена возможность нарушения их синтеза, причем, возникают патологические структуры, которые не способны расщепляться; в дальнейшем они откладываются в определенных тканях, как инородные тела. По-видимому, и в том и в другом случае имеется энэимопатия, нарушающая липоидный обмен.
Болезнь Гоше
(керазиновый ретикулоэндотелиоз)
Впервые эта болезнь была описана в 1882 г. Gaucher под названием изолированной первичной эпителиомы селезенки. Она развивается вследствие накопления цереброзидов в селезенке, печени и некоторых других органах. Болезнь Гоше встречается редко. В мировой литературе описано около 300 больных этой болезнью. В отечественной литературе описано только 32 больных (С. И. Рябов, 1961). Она наблюдается среди всех народов и с одинаковой частотой у мальчиков и девочек. Имеется два варианта болезни. Первый развивается хронически у старших детей и взрослых; это — локализованная форма. Второй вариант встречается исключительно в грудном возрасте и обусловлен неврологическими нарушениям и в центральной нервной системе.
Хроническая форма болезни Гоше, Болезнь начинается обычно в 5—8 лет, но может возникнуть и позже.
Клиническая картина. Одним из ранних симптомов болезни является увеличение живота вследствие значительной спленомегалии. Гипертрофия печени менее выражена и развивается позже. Желтуха и портальная гипертензия отсутствуют. Увеличение лимфоузлов наблюдается редко. Иногда обнаруживается местная припухлость в области бедер; дети при этом жалуются па спонтанные боли в нижних конечностях. Костные поражения бывают у больных не всегда, и большей частью они обнаруживаются только рентгенологически. Отмечается диффузный остеопороз пли сетчатое строение костной ткани; может наблюдаться также истончение кортикального слоя вследствие увеличения объема костного мозга. Если остеопороз носит ограниченный характер, возникают кистеобразные полости, обычно расположенные в верхней части бедра. В других случаях имеются явления остеосклероза. У некоторых больных обнаруживается деформация головки бедра, напоминающая асептический некроз. Вследствие деформации и уплотнения нижних отделов бедер, они принимают колбообразный вид, напоминая по форме «бутылку». Кроме того, могут наблюдаться по
ражения позвонков; обычно один иди два позвонка в поясничной области декальцинируются и уплощаются.
В крови можно обнаружить анемию, лейкопению, гранулопснию и тромбоцитопению. Количество липидов и холестерина в крови нормально.
Течение болезни. Общее состояние и вид ребенка могут длительно оставаться удовлетворительными, но в дальнейшем нередко отмечается отсталость физического развития. Далее после 10-летнего возраста может появиться бронзовый оттенок кожи лица, шеи. ладоней и стоп. Иногда эта пигментация становится диффузной и поражает также слизистые оболочки. В течение многих лет положение больного мало изменяется, затем состояние ухудшается, прогрессирует похудание, появляются боли в животе, кишечные расстройства, затруднения при мочеиспускании и отеки. В результате сдавления селезенкой области левой почки может развиться гидронефроз. Увеличиваются и костные изменения. В некоторых костных участках усиливается деструкция и вовлекаются в патологический процесс тазобедренный, коленный и голеностопный суставы. Позже могут появиться кровоизлияния и кровотечения вследствие выраженной тромбоцитопении. Анемия и лейкопения также прогрессируют.
Болезнь Гоше легче диагностируется, если имеется изолированная спленомегалия. При пункции костного мозга в мнелограмме обнаруживаются специфические клетки Гоше, которые имеют величину от 20 до 80 лк.м со светлой протоплазмой; ядро клетки — круглое или овальное, расположено эксцентрично; иногда в клетке имеется несколько ядер. Хроматин ядра располагается в виде нежной сеточки. В цитоплазме различают сет- чатость и фибриллярные нити, между которыми имеются большие лакуны, не воспринимающие обычную окраску. Такие же клетки можно обнаружить в спленограмме. Следует, однако, помнить, что пункция селезенки при болезни Гоше не всегда безопасна.
При дифференциальном диагнозе с тромбофлебитической селезенкой следует иметь в виду, что для нее характерны расширение вен пищевода и уменьшение размеров селезенки после кровотечений из желудка и пищевода; при циррозе печени — обычно имеется застой в области воротной вены. Иногда приходится
дифференцировать болезнь Гоше и с другими заболеваниями (хроническим лейкозом, лимфогранулематозом, ксантоматозом). В раннем детском возрасте большие трудности возникают при дифференциальной диагностике между острой формой болезни Гоше и болезнью Нимана — Пика. Прогрессирующее слабоумие более характерно для последнего заболевания.
При патологоанатомическом исследовании обнаруживают огромную селезенку, плотной консистенции. На разрезе видны нежные беловато-желтоватые пятна, следы старых и свежих геморрагий и небольшой склероз пульпы. При гистологическом исследовании находят много клеток Гоше в селезенке и печени; в последней они располагаются внутри долек, вблизи печеночных вен и в просвете разветвлений портальной вены. Характерен значительный аннулярный склероз. Лимфатические узлы брюшной полости тоже вовлечены в патологический процесс. В костях обнаруживаются деструктивные поражения и зоны некроза.
Химические исследования. Цереброзиды, выделенные из селезенки при болезни Гоше, представляют собою гликопереброзиды. У большинства больных выделяются гл и коцере б роз иды, а у некоторых — галактане реброзиды.
Патогенез. При болезни Гоше нормальные ретикулярные клетки фиксируют цереброзиды, в избытке циркулирующие в крови (Pick). Если животным впрыскивать цереброзиды в большом количестве, у них в селезенке появляются клетки, морфологически сходные с клетками Гоше. Однако после прекращения инъекций эти клетки исчезали. По данным Thannhanser, отложения цереброзндов наступают в результате нарушения внутриклеточного обмена, поэтому в сыворотке больных цереброзиды не обнаруживаются, а эритроциты содержат такое же их количество, как и у здоровых людей (0,2 jna%}. Предполагается, что имеется снижение активности фермента цереброзидазы. Исследования Radin с соавт. показали, что галактоза, меченная C14, быстро внедряется в состав цереброзндов мозга. По Любаршу ретикулярные клетки вилочковой железы, надпочечников, поджелудочной железы, перибропхиалыюй и периваскулярпой ткани легких теряют способность накапливать липоидные вещества 54
і концу первого года жизни. В связи с этим у старших детей в легких не находят клеток Гоше (Rath).
Наследственность. При генетическом исследовании ‘250 больных с болезнью Гоше у 89 обнаружена семейственность этого заболевания. Изредка можно отметить болезнь Гоше у детей и их родителей. Кровное родство родителей наблюдается исключительно редко. По В. П. Эфронмсону, Herndon и Bender наследственность носит аутосомный и рецессивный характер, однако Groen относит наследственность при болезни Гоше к доминантным. Возможно, что при этой болезни имеются два генетических варианта наследования. Интересно, что острые и хронические формы в одной и той же семье не встречаются.
Лечение. При развитии анемии назначают железо, трансфузии крови, витамины. При болях в костях применяют местную рентгенотерапию. У некоторых больных приходится прибегать к спленэктомии. Показанием к оперативному вмешательству служат геморрагические явления вследствие гиперспленизма. Спленэктомия, кроме того, показана при нарушении функции кишечника и мочевыводящнх органов вследствие сдавления нх огромной селезенкой. После операции улучшается общее состояние больных; количество эритроцитов, лейкоцитов и тромбоцитов увеличивается и прекращается кровоточивость; исчезает и характерная бронзовая окраска кожи. Однако спленэктомия не излечивает болезни Гоше; инфильтрация печени и костного мозга специфическими клетками продолжается, причем размеры печени настолько увеличиваются, чго могут вести к значительному сдавлению оргаиоз брюшной полости. Смерть наступает в 20—30 лет от прогрессирующей кахексии, интеркуррентной инфекции или от кровоизлияния в жизненно важные органы.
Острая форма болезни Гоше. В настоящее время в литературе описано свыше 30 больных острой формой (Giampalnio). Заболевание наблюдается исключительно в раннем грудном возрасте. Уже при рождении отмечается повышенная сонливость детей. При обследовании можно обнаружить значительное увеличение селезенки и менее выраженное увеличение печени. Общие расстройства выявляются быстро. Наступает похудание и лихорадка, На первый план выступают
псрішьіе расстройства; нарушение сознания, выраженная заторможенность, апатия, моторные нарушения, мышечная гипертония, опистотопус и расстройство дыхания, сопровождающееся цианозом. Нередко наступают расстройства глотания, косоглазие и слепота. Иногда развиваются параличи с различной локализацией. Все эти симптомы связаны с прогрессирующей декортикацией.
Течение болезни обычно быстрое; смерть наступает через 3—6 мес, от начала болезни и нередко обусловлена вторичной интеркуррентной инфекцией. У некоторых больных болезнь может протекать подостро.
Патологоанатомические изменения. При острой форме часто имеется инфильтрация легких, состоящая из клеток Гоше и занимающая альвеолы и перегородки, а также отмечаются поражения почек, надпочечников, вилочковой железы и лимфоидной ткани кишечника. Пигментные аномалии отсутствуют. Поражение мозга встречается только при острой форме. В больших и малых пирамидальных клетках коры головного мозга, в особенности в париетально-окципитальной области можно обнаружить много вакуолей. Клетки растянуты и мумифицированы. Клеточные вакуоли не окрашиваются красками, окрашивающими жир. В мозгу обычно не находят клеток Гоше. Изредка их находит в периваскулярной ткани мозга.
Мы наблюдали в клинике проф. А. Ф. Тура больную хронической формой болезни Гоше, краткие сведения о которой считаем полезным привести.
Больная А. Э., 11 лет, поступила п клинику 31 октября 1964 г. Родилась в срок с весом 2600. Период ново рожден пости и грудной возраст протекали нормально. В дальнейшем перенесла корь, краснуху, ветряную оспу и бронхопневмонию На втором году жизни родители заметили увеличение живота, но не придали этому значения. Около четырех лет общее состояние ребенка несколько ухудшилось; увеличился больше живот, понизился аппетит, появились носовые кровотечения π стала заметной отсталость физического развития. В 7 лет впервые при врачебном осмотре было обнаружено увеличение селезенки до уровня пупка и печени — на 5 см ниже реберной дуги. 10/lX 1957 г. в хирургическом отделении ЛПМИ больной была успешно произведена спленэктомия. Вес селезенки был равен 756 г. При гистологическом исследовании среза селезенки обнаружены клетки Гоше в значительном количестве. При осмотре в 1964 г. получены следующие патологические данные. Больная значительно отстала в физическом развитии. По антропометрическим
никпзателям девочка соответствует C1A годам. Бледность кож-
....... покровов и видимых слизистых оболочек. IIa лице много
пгснушек; отдельные пигментные округлые пятна на туловище. :■ тле и венозный р псу но к на груди и передней стенке живота.
1 Ia коже нижней челюсти сосудистые «звездочки». Резкое увеличение живота. При пальпации живота прощупывается огром- IHiH плотная гладкая безболезненная печень, которая занимает ∙,∕ι полости живота. Нижний край ее спускается в малый таз. Органы грудной клетки без особенностей. Больная жалуется на боль в верхней трети правого бедра, пальпация этой области болезненна. Походка напоминает утиную. На рентгенограммах тазобедренных суставов и бедер определяется заметное разрежение костной структуры по разлитому типу. Остеопороз в области шейки бедер больше выражен справа. В нижних отделах бедер — утолщение метафнзов (колбообразный вид нижней трети бедер). В пунктате костного мозга и печени обнаружено много одно- и двухъядерных клеток Гоше. Свертывающая система крови в норме, количество тромбоцитов до операции в 1957 г. 46 970 —в 1 ΛΛa, в 1964 г.— 316 720. Количество холестерина 170 нг%, билирубина 0,25 мг%, реакция непрямая. Общий белок 6,7%, диенротеинемия — А/Г = 1,3. Лейкоцитов — 12 200, лейкоцитарная формула и пределах возрастной нормы, РОЭ — 30 мм в 1 ч. Лечение симптоматическое.
Семенная амавротическая идиотия
(болезнь Тей—Сакса)
При семейной амавротической идиотии изменения со стороны глаз впервые были описаны офтальмологом Tay в 1881 г. Вскоре, в 1887 г,, английский невропатолог Sachs дал подробное описание клинической картины и патологоанатомических изменений центральной нервной системы, характерных для этого заболевания. Ои первый назвал болезнь семейной амавротической идиотией.
Клиническая картина. Заболевание встречается преимущественно в семьях с кровным родством. Мальчики и девочки поражаются с одинаковой частотой. Для больных детей характерно ожирение в первые месяцы жизни (С, Г. Берлянд и С. Л. Левин). К характерным симптомам болезни относится отсутствие интереса к окружающему и прогрессирующая мышеч пап слабость. Постепенно больной утрачивает приобретенные до того навыки. Движения становятся более слабыми и редкими. Он не способен сидеть, удерживать игрушки и держать голову. Дальнейшее прогрессирование болезни приводит к тому, что ребенок
лишается способности менять свое положение и лежит без движений, совершенно инертный. Столь же постепенно слабеет зрение, больной перестает различать предметы, затем наступает полная слепота, вследствие атрофии зрительных нервов. Иногда наблюдается ги- перакузия, из-за которой больные становятся чувствя- тельными к малейшему шуму. Через несколько месяцев гипотония мышц сменяется стойкой гипертонией, имеющей характер децеребрационной ригидности. Сухожильные рефлексы обычно повышены, но могут быть и пониженными. Патологические рефлексы встречаются редко. К непостоянным симптомам амавротической идиотии относятся косоглазие, нистагм, гиперкинезы, повышение возбудимости периферических нервов и глухота. Изредка наблюдаются эпилептические припадки, которые обычно протекают абортивно без клонической фазы судорог (С. С. Мнухин, Marchand с соавт.). При клиническом обследовании обнаруживаются в основном только неврологические расстройства. Со стороны внутренних органов отклонений от нормы не находят. Печень и селезенка не увеличиваются. В период развития заболевания отмечается анорексия, склонность к запорам и прогрессирующая кахексия. У некоторых больных па коже могут появляться подушкообразные утолщения и желто-канареечная пигментация на кистях, стопах и бедрах. Характерны симптомы со стороны глаз. В области macula Iuiea появляется вишнево-красное пятно, вокруг которого имеется беловатая блестящая зона. Кроме того, постепенно выявляются изменения в окраске зрительных сосков, свидетельствующие об атрофии зрительных нервов. Сосуды сетчатки остаются нормальными. Изменения в области желтого пятна обычно мало меняются в течении болезни. В некоторых случаях офтальмологические поражения носят атипичный характер и характерное вишнево-красное пятно может отсутствовать.
На электроэнцефалограмме имеются замедленная дисритмия с увеличенными зубцами н атипичными волнами. В течение болезни развивается глубокое слабоумие и полный маразм. Кормление детей становится очень затруднительным. Ребенок погибает обыкновенно через 1 — 1 ,∣2 года от начала болезни, однако
некоторые больные достигают 4—5-летнего возраста. При этом может изменяться картина глазного дна: исчезают изменения в области желтого пятна и остается только атрофия зрительных нервов. Впоследствии может присоединиться пигментный ретинит.
Патогенез. Предполагается, что основная роль принадлежит нарушению внутриклеточного обмена; по- видимому, нарушается катаболизм ганглиозидов. Кроме ранней детской формы амавротической идиотии, различают еще позднюю детскую форму. Для нее характерно появление заболевания в возрасте 2—4 года. Основными симптомами поздней формы являются понижение зрения, постепенно приводящее к слепоте ΓΙ прогрессирующее слабоумие; кроме того, иногда присоединяется мозжечковая атаксия. Течение заболевания такое же, как при ранней форме, В течение нескольких лет развивается кахексия и маразм, приводящие больного к смерти. Поражение глаз отличается от изменении при ранней детской форме. Значительно реже наблюдается вишнево-красное пятно. Чаще отмечается пигментный ретинит или изолированная атрофия зрительных нервов. C помощью электроретиногра- фии удается выявить дегенерацию невроэпителия, которая офтальмоскопически не распознается.
Врожденная форма. Norman и Alood в 1941 г. описали особую форму амавротической идиотии у девочки, умершей па 17-день жизни. В 1954 г. Brown с сотр. наблюдал у младшей сестры этой умершей девочки амавротическую идиотию, приведшую ее к смерти в возрасте 7 недель. При офтальмоскопии была обнаружена двусторонняя атрофия зрительных нервов и почти полное отсутствие сосудов сетчатки. При врожденной форме больные отличаются не только врожденным характером заболевания, но и иной природой гистохимических изменении. При этой форме амавротической идиотии обнаружены значительные скопления холестерола в белом веществе мозга. В клетках печени, селезенки, надпочечников и лимфатических узлов найдено увеличенное количество липидов.
Юношеская форма амавротической идиотии была описана в 1903 г. Batten. Sjogren в 1931 г. уточнил симптоматологию и наследственный способ передачи юношеской формы амавротической идиотии потомству.
Ребенок рождается внешне здоровым и первые годы жизни хорошо развивается. Заболевание возникает между пятью и восемью годами. Первым сипмто- мом болезни является понижение зрения. Оно постепенно прогрессирует и заканчивается полной слепотой. Одновременно выявляется умственная отсталость, которая впоследствии доходит до глубокою слабоумия. Довольно рано могут появляться судороги. Позже развиваются другие неврологические расстройства. Вначале они носят характер экстрапнрамидных симптомов с ригидностью и скованностью движении, препятствующих в дальнейшем ходьбе. Позже появляются мозжечковые cHM∏τo1Mbi и особенно часто наблюдается нарушение равновесия, В конце концов развивается атаксия и гипертония мышц, которые препятствуют движениям больного. Постепенно общее состояние значительно ухудшается и смерть обычно наступает к 19 годам. Поражение глаз при юношеской форме отличается от изменений, которые обнаруживаются при детской форме амавротической идиотии. Вишнево-красное пятно отсутствует; явления дегенерации выражаются в пигментном ретините и хореоре- тините, которые иногда сопровождаются атрофией зрительных нервов. Изменения со стороны глаз неуклонно прогрессируют. Sjogren различает 5 этапов в течении юношеской формы заболевания: 1) глазные поражения, 2) судороги, 3) экстрапнрамидные признаки, 4) умственная отсталость с расстройствами речи и мозжечковыми нарушениями и 5) терминальный период с глубоким слабоумием, спастической тет- раплегией и полным маразмом.
Для клинической картины имеет значение, в каком возрасте возникает амавротическая идиотия. Чем раньше начинается заболевание, тем скорее оно прогрессирует и заканчивается летально.
Для амавротической идиотии наиболее характерными являются иатологоанатомивеские изменения нейтральной нервной системы. Ганглиозные клетки набухают и принимают овальную или грушевидную форму. Ядра клеток всегда располагаются эксцентрично и часто имеют пи цноти ческий вид. Цитоплазма содержит нежные липоидные включения и вакуоли. Тельца Ннссля разрушаются. Дендриты тоже деформируются
и набухают. Указанные изменения можно обнаружить и различных частях нервной системы. Клетки содержат в большом количестве вещество, которое окрашивается гематоксилином и липоидными красками в черный цвет. Кроме клеточных изменений, наблюдается демиелинизация нервных стволов и пролиферация невроглии со значительным увеличением числа звездчатых клеток. При ранних формах поражения центральной нервной системы имеют наибольшее распространение. При поздних формах они косят более ограниченный характер.
Следует различать два типа поражения сетчатки глаз: при первом имеется дегенерация ганглиозных клеток, при втором дегенерация зрительного эпителия с разрушением клеток. Этот процесс часто сопровождается пролиферацией глии и реакцией пигментного эпителия. При ранней детской форме болезни дегенерация сетчатки является изолированной и сопровождается уменьшением числа ганглиозных клеток и атрофией зрительных нервов. В более старшем возрасте, при юношеских формах, а иногда и при ранних детских — опа может отсутствовать.
Патологоанатомические изменения сводятся к вакуолизации клеток печени, селезенки, почек, легких и лимфоцитов. Следует отметить, что по окраске эти клетки отличаются от клеток нервной системы. Вакуолизацию клеток иногда можно обнаружить и в ретикулоэндотелиальной системе. Содержание ганглиозидоа в сером веществе мозга при амавротической идиотин значительно превышает их количество у здоровых детей (Cumings, Svenerholm). По Klenk, содержание ганглиозидов у больных в 5—10 раз выше, чем в норме; причем, структура ганглиозидов качественно ничем не отличается от нормальной. Увеличение ганг- лиозндов идет за счет содержащих гексозамнны. При ювенильных и поздних детских формах ганглнозиды не увеличиваются в сером веществе мозга, а в белом веществе они совсем отсутствуют. По нарушению ли погідного обмена и прогрессирующему течению в раннем детском возрасте амавротическая идиотия имеет сходство с болезнью Нимана — Пика. При дифференциальной диагностике следует иметь в виду, что при
последнем заболевании имеется увеличение печени и селезенки и гистохимически определяется увеличение сфингомиелина, а при амавротической идиотии их количество нормально.
Генетические данные. В литературе имеется описание заболевания амавротической идиотией у однояйцевых близнецов. Большинство исследователей отмечает аутосомный рецессивный характер наследственной передачи. Действительно, больные чаще всего рождаются от здоровых родителей. Кровное родство родителей при амавротической идиотии встречается нередко и в различных комбинациях: брат и сестра (у нашего больного), двоюродные братья и сестры (С. Т. Берлянд н C. Л. Левин, Богаерт и др.), дядя а племянница (А. Фельдман). Заболевание часто наблюдается у нескольких членов семьи. По данным Falkenlieim, в 13 семьях, изученных разными врачами, амавротическая идиотия была обнаружена у 37 детей (от 2 до 5 больных в каждой семье). Sjogren обнаружил в Швеции 115 заболеваний юношеской формой в 59 семьях. Кровное родство родителей отмечалось в 25,4% этих семей. По данным Klein и Klenides, кронное родство родителей встречается при детской форме в среднем в 24,5—27,6%. В одной и той же семье не бывает чередования детской и юношеской формы болезни. Однако нередко в семьях наблюдается заболевание детской и поздней детской формой; кроме того, описаны семьи, в которых наряду с юношеской формой имелись больные поздней детской формой амавротической идиотии.
Современное лечение не в состоянии изменить трагического течения болезни Тей — Сакса. В качестве иллюстрации приводим собственное наблюдение.
Больная Н. И,, I1A лет, привезена из Нарьян-Мара Архангельской области Родители — родные брат и сестра. Отцу 20 лет, матери 14 лет. Девочка с З’Д мес. воспитывается в Доме ребенка. Родилась доношенной с весом 3 кг и ростом 48 си Период новорожденностн протекал нормально. Развивалась а Доме ребенка замедленно. Состояние девочки ухудшилось с 9 мес. после перенесенной ветряной оспы. Стала отставать в росте и весе; появились срыгивааия и анорексия. Значительно снизился тонус мышц. В 1 год 2 мес. появилось сходящееся косотлазче. Первые зубы прорезались в G мес. Самостоятельно сидела с 1 год 2 мес.
Стояла с 1 года J мес., но после 1 года 3 мес. постепенно утратила эти навыки Тургор тканей стал дряблым, развилась гипотрофия I! степени, прогрессирующая гипотония мышц. При осмотре получены следующие
данные: общее физическое развитие соответствует развитию 7-месячного ребенка. Лежит в кровати преимущественно неподвижно (рнс. 1). Легкий тремор рук. На прикосновение н легкий массаж реагирует плачем. Голову не держит. При вдохе — втяжекие межрсберий.
Со стороны внутренних органов — без особенностей. Печень мягкая, прощупывается на 3 см ниже реберной дуги, селезенка не прощупывается.
Коленные рефлексы ослаблены, ахилловы — отсутствуют. Отсталость психомоторного и психического развития. Офталь- мо-скопически — атрофия зрительных нервов обоих глаз.
Область желтого пятка не изменена. Сходящее косоглазие.

Рис. 1. Вольная Н. И. Болєзріь Тей—Сакса.
Лабораторные исследования крови, мочи, кала в пределах нормы. RW — отрицательна. Мнелограмма— нормальная. Рентгенограмма костей — без особенностей. Состояние больной прогрессивно ухудшалось. За 2 мес. потеряла в весе 800 г. Гипотония нижних конечностей через C недель сменилась гипертонией. Симптоматическое лечение без эффекта. Выписана в Нарьян-Марский Дом ребепка по просьбе родных.
Прогрессирующая липодистрофия
(болезнь Варракера — Симонса,
Lipodistrophia CephaIothoracica progressiva)
Прогрессирующая липодистрофия относится к самым древним заболеваниям. Известно, что живший в XlV веке до нашей эры египетский фараон Эхнатон страдал с детства прогрессирующей липодистрофией. Впервые заболевание описано в начале ΧλΠΙΙ века Morgagni. Подробно болезнь описал Barraquer в
2 А. М. ⅛6β3raya
S3
1906 г. Прогрессирующая лнподпстрофия— редкое заболевание; Jeun с соавт. в мировой литературе нашли описание 224 больных. В отечественной литературе описание таких больных не превышает 20. Женщины болеют в 3—4 раза чаще, чем мужчины.
По данным Oppermann и соавт., прогрессирующая липодистрофия начинается в 84% между 5 и 15 годами жизни больных.
Н. Б. Беленькая и другие авторы считают, что пубертатный период благоприятствует развитию болезни. Сущность заболевания заключается в постепенном безболезненном исчезновении подкожно-жирового слоя на голове, лице, шее, туловище и руках. В противоположность этому нижняя часть тела имеет нормальную или гипертрофированную жировую клетчатку. Различают три клинические формы прогрессирующей лнподистрофии: 1) сочетание липодистрофии верхней половины тела с липогипертрофией нижней; 2) липодистрофия верхней половины туловища при нормальной нижней и 3) нижняя липогипотрофия. У детей чаще имеется вторая форма — изолированной верхней липодистрофии.
Этиология и патогенез окончательно не выяснены. Высказывались предположения о возможной роли наследственного предрасположения и влияния некоторых внешних факторов. Имеют значение в возникновении болезни психические травмы, энцефалит, инфекционные болезни и местные инфекционные очаги. «Эндокринный» патогенез заболевания не подтвердился.
Наиболее вероятной гипотезой является поражение гипофизарно-диэнцефальной области. В пользу такого предположения говорит важная роль, которую играет гипоталамус в жировом обмене и обнаружение анатомических изменений в этой области у некоторых больных прогрессирующей липодистрофией (Sandmann и др.). Описаны при этом заболевании опухоли межуточного мозга, кальцинаты выше турецкого седла и другие поражения.
Клиническая картина. Начало болезни незаметное, постепенное. Заболевание начинается с атрофии
подкожного жира на лице, которое становится маскообразным. Поражение обычно бывает симметричным, причем характерно исчезновение и комочков Биша, которые сохраняются у детей при других видах дистрофии. Иногда атрофии подкожно-жирового слоя предшествует отечность лица и груди. Далее постепенно исчезает подкожно-жировой слой на голове, шее, грудной клетке и руках. В нижней части тела атрофического процесса не наблюдается, так что создается впечатление, что жир из верхних частей тела опустился в нижележащие отделы. Исчезновение жировой клетчатки происходит в течение Р/г—5 лет и в любой момент может остановиться. Пораженные части тела остаются и а всю жизнь атрофичными и отложения в них жира пе происходит. Мышечная система на местах липодистрофии остается нормальной. Электровозбудимость мышц не претерпевает изменений. Иногда отмечаются хореатетозные движения. Наблюдаются также трофические расстройства кожи, волос, ногтей, гипертрихоз и гирсутизм. Иногда отмечается повышенная потливость при сухости кожи в подмышечных областях и усиленная секреция сальных желез. Могут отмечаться и психические нарушения со склонностью больных к депрессии. Больные жалуются на повышенную возбудимость, вялость, слабость, раздражительность и зябкость. Со стороны желудочно-кишечного тракта отмечается нередко анорексия, запоры, иногда поносы, а со стороны сердца бывают функциональные нарушения. У tA больных обнаруживаются изменения со стороны почек в виде пиелонефрита, гломерулонефрита или аномалий развития мочевыделительной системы. Описано сочетание прогрессирующей липодистрофии с сахарным диабетом и с тиреотоксикозом. Встречается и несахарны и диабет, однако эндокринные нарушения носят вторичный характер.
При лабораторных исследованиях иногда можно обнаружить лимфоцитов и эозинофилию, а также повышение холестерина в крови, гиперлипемию и повышение активности липазы. Часто наблюдается тран- зиторная альбуминурия и диспротеииемия. Дифференциальный диагноз может потребоваться при дистрофии вследствие anorexia nervosa. Различие заклю-

Рис. 2. Больной С. А. Прогрессн рующая липодистрофия. ·
чается в том, что при прогрессивной лигюдистрофии истощение распространяется только на верхнюю половину туловища, а при anorexia nervosa дистрофия распространяется равномерно. Кроме того, при последнем заболевании сохраняются комочки Биша на лине.
Лечение прогрессивной липодистрофии представляет собою весьма неблагодарную задачу. В последнее время рекомендуется комплексное лечение анаболическими и катаболическими гормонами, инсулином, препаратами гипофиза и щитовидной железы. Применяется также рентгенотерапия гипоталамической области. Больные принуждены также прибегать к хирургической помощи для производства пластической операции (трансплантация жировой ткани, перлоно- вая пломба). К сожалению, через некоторое время наступает рассасывание трансплантата.
Прогноз для жизни благоприятный, но восстановления подкожно-жирового слоя в верхних отделах тела не происходит в течение всей жизни больного.
Приводим собственное наблюдение.
Больной С. А., 7l∕s лет поступил в клинику 5/IIl 1968 г. с жалобами на резкое похудание липа, головокружения и преходящие боли в животе. Родился от первой беременности с весом 2800 а и длиной тела 50 сл. Развивался и раннем детстве вполне удовлетворительно. C 5'∕j лет отмечено понижение аппетита, стад худеть.
При осмотре общее состояние удовлетворительное. Резкое истощение лица и шен. Кожа здесь морщинистая, комочки Бнша отсутствуют. Ma туловище подкожно-жпровой слой слегка истончен, а на нижипх конечностях — нормален (рис. 2). Рост 125 с.н, вес 21 кг, Над областью сердца систолический шум функционального характера, в остальном внутренние органы в пределах возрастной нормы.
При биохимическом исследовании крови К. Ca. Cl, NaCl, белковые фракции, билирубин крови, сахар оказались нормальными. Содержание холестерика понижено — 90 жг%. Результаты морфологического исследования крови также в норме. Функция почек нормальна. Рентгенологическое исследование грудной клетки и костей без патологических изменений. При внутривенной урогра- фни обнаружена умеренная пнелоэктазня и расширение чашечек левой почки.
В клинике установлен диагноз прогрессирующей лнподнстро-
фии.
Лечение анаболическими и катаболическими гормонами оказалось безуспешным, дистрофические явления на лице и шее остались без перемен *.
Аналогичный случай мы наблюдали в областной клинической больнице.
Еще по теме Фtнилпнровиногра дная олигофрения (фенилжетонурия):
- 13.3. Паранойя, олигофрения, психопатия
- Отграничение вторичной задержки умственного развития при нарушениях речи от олигофрении и ЗМР.
- Особенности развития познавательной сферы и ведущих видов деятельности при олигофрении
- Умственная отсталость: определения, классификация. Психологическая структура дефекта при олигофрении и деменции.
- 16. Общее стойкое психическое недоразвитие как вариант психического дизонтогенеза.
- Содержание
- 2. Классификации ЗПР
- Синдром клайнфельтера
- Глава I НАСЛЕДСТВЕННЫЕ ЭНЗИМОПАТИИ
- 17. Поврежденное развитие как вариант психического дизонтогенеза.
- ХI.1. УМСТВЕННАЯ ОТСТАЛОСТЬ