Основы патогенеза анемии при хронических болезнях
Цитокины и клетки ретикулоэндотелиальной системы вызывают изменения в гомеостазе железа, пролиферации эритроидных клеток-предшественников, продукции эритропоэтина, продолжительности жизни эритроцитов, каждое из которых вносит вклад в патогенез анемии [131,248,252].
Типичным представителем «анемии воспаления», обусловленной действием исключительно цитокинов является анемия, ассоциированная с ревматоидным
артритом, известная как ревматоидная анемия [149]. При отсутствии эффективного лечения анемия широко распространена среди пациентов с ревматоидным артритом (по данным различных исследований от 33,3 до 59,1%) [185].
Нарушение регуляции гомеостаза железа в организме
Отличительной особенностью АХБ является развитие нарушений гомеостаза железа с повышенным потреблением и накоплением железа в клетках ретикулоэндотелиальной системы. Это приводит к перенаправлению железа из циркуляции в хранилище в ретикулоэндотелиальной системе. С последующим ограничением доступности железа для эритроидных клеток-предшественников и, как следствие, железодефицитному эритропоэзу [43,245].
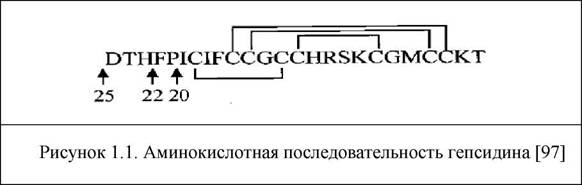
В 2001 году впервые были опубликованы данные о выделении из мочи человека нового пептида, который, как полагали, обладает антимикробными свойствами, так как богат цистеином. Название белка гепсидин (hepcidin) было образовано от: hepar (лат. - печень - место синтеза пептида) и cidin (лат. - уничтожитель - подчеркивает антимикробные свойства пептида) (рисунок 1.1.) [97]. Позднее было показано, что синтез гепсидина происходит и в тубулярном аппарате почек, и в других клетках, тканях и органах, включая макрофаги, адипоциты и клетки головного мозга, что может говорить о важной роли гепсидина в аутокринном и паракринном контроле обмена железа на локальном уровне [73,1б2,221,255]. Выделение гепсидина - острофазного белка-регулятора обмена железа, состоящего из 25 аминокислот - помогло пролить свет на связь иммунного ответа и гомеостаз железа и АХБ [240].
Гепсидин кодируется как 84-аминокислотный препропептид [237]. Зрелый гормон циркулирует в плазме и связан с а2-макроглобулином [98]. В то время как активирующее действие этой связи было описано, механизмы выведения гепсидина до сих пор до конца неизвестны. Основной путь выведения гепсидина - его экскреция с мочой. Когда функция почек не нарушена, концентрация гепсидина в моче коррелирует с уровнем циркулирующего гепсидина, без видимой регуляции процесса экскреции. Однако, основываясь на сопоставлении между концентрацией гепсидина в сыворотке крови и моче, только 5% гепсидина плазмы фильтрующегося почками, выводятся в неизмененном виде с мочой. Это говорит о том, что гепсидин не может свободно фильтроваться в клубочках и/или отфильтрованный гепсидин реабсорбируется и деградирует в проксимальных канальцах, аналогично другим маленьким пептидным гормонам [109,231]. Гепсидин также может быть выведен рецептор-опосредованным эндоцитозом в тканях, экспрессирующих его рецептор - ферропортин, что показывает накопление меченого гепсидина в тканях богатых ферропортином и разрушением путем эндоцитоза комплекса гепсидин-ферропортин в культивируемых тканях [213].
По своей структуре гепсидин напоминает изогнутую шпильку для волос, сцепленную между собой четырьмя дисульфидными мостиками. Последние данные показывают, что для связи гепсидина с рецептором требуется участие одной из дисульфидных связей. Удаление одной из дисульфидных связей не приводит к значимому снижению активности гепсидина in vitro, это свидетельствует о том, что множественные дисульфидные связи могут способствовать формированию контакта с ферропортином [223].
Амфипатическая структура гепсидина и его множественные дисульфидные связи обычно характеризуют антимикробные и антигрибковые пептиды. Однако гепсидин демонстрирует лишь скромные антимикробные свойства in vitro и только в больших концентрациях (10 - 30 ііМ), значимость его антимикробных возможностей in vivo не определена.
У пациентов с наследственнымгемохроматозом, связанным с дефицитом гепсидина, описано развитие инфекций, вызванных атипичными микроорганизмами (Vibrio, Yersinia и Listeria). Но эта восприимчивость может быть связана с преимуществами, полученными бактериями, в связи с повышенным уровнем железа, более чем с потерей непосредственно антибактериального эффекта гепсидина [162]. Структура гепсидина представлена на рисунке 1.2.
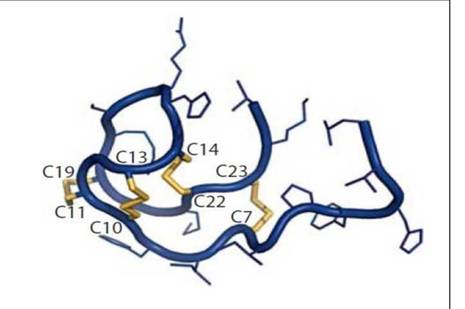
Рисунок 1.2. Строение гепсидина [162]
Гепсидин является главным регулятором концентрации железа в плазме. Инъекция гепсидина мышам приводит к значительному снижению сывороточного железа уже в течение часа. Несмотря на то, что гепсидин быстро выводится из плазмы, эффект от введения одной дозы сохраняется до 72 часов. Вероятно, это время требуется для ресинтеза достаточного количества рецептора гепсидина - ферропортина [94,213].
Воспаление у гепсидин-дефицитных мышей не приводит к снижению железа сыворотки [53,107]. Индукция гипоферремии интерлейкином-6 и гепсидином развивается в течение нескольких часов и не отмечается у мышей с выключенной
экспрессией интерлейкина-6, обработанных скипидаром (как модель воспаления) [107].
Хроническая гиперпродукция гепсидина приводит к развитию железоограниченной анемии у мышей и человека [204]. Напротив, дефицит гепсидина приводит к перегрузке железом с накоплением железа в печени и других органах. Полное отсутствие гепсидина приводит к развитию ювенильного гемохроматоза, наиболее тяжелой формы наследственного гемохроматоза [138,145,222].
Проявления избытка и дефицита гепсидина показывают, что гепсидин ингибирует всасывание железа в кишечнике и представление железа из макрофагов, утилизирующих старые эритроциты [71]. При гиперпродукции гепсидина в период эмбрионального развития у плода развивается железодефицитная анемия и многие умирают при рождении, это свидетельствует о том, что гепсидин ингибирует трансплацентарный транспорт железа [204].
Гепсидин, видимо, также блокирует, по крайней мере частично, экспорт запасов железа из гепатоцитов, на это указывает накопление железа в печени у мышей, страдающих гепсидинпродуцирующими опухолями [90]. Гепсидин, произведенный вне печени, может осуществлять контроль над локальным обменом железа в тканях, в которых он продуцируется. Например, центральная нервная система отделена от плазмы гематоэнцефалическим барьером и циркулирующий гепсидин не может быть транспортирован через этот барьер. Однако было описано, что ткани мозга могут сами продуцировать гепсидин, сохраняя возможность регулировать местный метаболизм железа независимо от системного контроля [196,253].Гепсидин действует, модулируя экспорт клеточного железа в плазму и внеклеточную жидкость, посредством ферропортина. Ферропортин - это одновременно и рецептор гепсидина и единственный известный экспортер клеточного железа у позвоночных. Ферропортин экспрессируется на клетках, являющихся профессиональными обработчиками железа в организме: энтероцитах двенадцатиперстной кишки, абсорбирующих пищевое железо,
макрофагах печени и селезенки, утилизирующих старые эритроциты, гепатоцитах, запасающих железо и трофобластах, транспортирующих железо плоду во время беременности [220]. Ферропортин также экспрессируется на эритроидных клетках предшественницах, это позволяет предположить, что его присутствие позволяет повысить чувствительность клеток-предшественниц к системному уровню железа и помогает определить направление их роста и дифференцировки [7]. Было показано, что полное отсутствие экспрессии ферропортина у мышей приводило к смерти эмбрионов, в связи с невозможностью эмбриональных трофобластов транспортировать железо от матери к плоду. У мышей с селективной блокировкой продукции ферропортина (с сохраненным плацентарным ферропортином) новорожденные мыши, лишенные ферропортина, имели тяжелую железодефицитную анемию, вследствие низкого всасывания пищевого железа в 12-ти перстной кишке, а также нарушенного представления железа из печеночных запасов и макрофагов, утилизирующих железо [220].
Посттрансляционнный контроль уровня ферропортина его лигандом гепсидином - ведущий способ регуляции ферропортина. Связывание гепсидина с ферропортином запускает интернализацию и деградацию комплекса лиганд- рецептор [94]. Связывание, вероятнее всего, включает в себя дисульфидный обмен между одной из дисульфидных связей гепсидина и наружным тиольным остатком Cys326 ферропортина. У пациентов с мутацией Cys326S развивается рано выявляемая перегрузка железом, мутантный ферропортин теряет способность связываться с гепсидином in vitro. Как только произошла интернализация, комплекс гепсидин-ферропортин деградирует в лизосомах и транспорт клеточного железа прекращается. Экспрессия гепсидина может также регулироваться независимо от гепсидина, клеточным содержанием железа. Механизм транспорта железа ферропортином остается неисследованным [7].
Гепсидин гомеостатически регулируется уровнем железа, гемопоэтической активностью и гипоксией. Избыток железа стимулирует продукцию гепсидина и
повышение гормона, в свою очередь, блокирует всасывание железа в кишечнике, предотвращая дальнейшую нагрузку железом. Напротив, продукция гепсидина угнетается при дефиците железа, позволяя повысить всасывание пищевого железа и пополнение запасов железа. Повышение эритропоэтической активности также оказывает супрессивный эффект на продукцию гепсидина. Помимо повышения всасывания железа это позволяет быстро предоставить запасы железа из макрофагов и гепатоцитов и увеличить доставку железа для эритропоэза. Кроме того, гипоксия снижает продукцию гепсидина, вызывая эффекты, описанные выше. Молекулярный механизм, лежащий в основе регуляции уровня гепсидина посредством уровня железа и активностью эритропоэза, является областью интенсивных исследований, но все еще не полностью понят. Уровень гепсидина также увеличивается при воспалении и инфекции, и это предполагает, что данный механизм является ответом иммунной системы с целью ограничения доступности железа для микроорганизмов. Считается, что воспаление является наиболее мощным регулятором продукции гепсидина и подавляет действие других механизмов [33,68,93,116,147,227,254].
Регуляция продукции гепсидина опосредованная железом осуществляется, вероятнее всего, совместно циркулирующим железом, связанным с трансферрином (FeTf), и клеточными запасами железа. Кроме того, железосенсорные молекулы для внеклеточного и внутриклеточного железа, видимо, разные, но они, вероятнее всего, используют путь костномозгового морфогенетического протеина (BMP) для повышения экспрессии гепсидина. Данный путь возник как крайне необходимый регулятор экспрессии гепсидина [9]. Обычно, корецептором в данном процессе является гемоювелин (HJV). У людей с мутацией, разрушающей HJV развивается перегрузка железом, связанная с удалением гепсидина, без других видимых нарушений [158,165]. Второй путь регуляции продукции гепсидина включает в себя трансферриновые рецепторы 2 (TfR2) и HFE. TfR2 являются гомологами TfR1 - рецепторами, необходимыми для захвата железа энтероцитами, также экспрессируется на многих других типах клеток [80,226]. HFE - протеин аналогичный MHC 1 класса. Он не связывает
железо, но влияет на метаболизм железа опосредованно через железосвязывающие белки - TfR1 и TfR2. В исследованиях было показано, что взаимодействие HFE и TfR2 необходимо для индукции гепсидина в ответ на FeTf [118]. Мутации HFE и TfR2 описаны при взрослой форме наследственного гемохроматоза. Также было описано взаимодействие HFE и TfR2 с HJV и возможно, что связывание FeTf с TfR1 и TfR2 инициирует формирование суперкомплекса состоящего из HFE, TfR2, HJV и BMP рецепторов [133]. Помимо FeTf, гепсидин может также регулироваться запасами железа, предположительно некоторыми формами внутриклеточного железа. Механизм участия внутриклеточного железа в регуляции гепсидина пока остается неясным, однако в последнее время на первые позиции в этом процессе вышел BMP6, который действует, связываясь непосредственно с HJV. Однако еще предстоит определить, как продукция BMP6 регулируется железом [32]. Также было описано наличие молекул, взаимодействующих с HJV и изменяющих его экспрессию на поверхности клеток. Это протеаза - матриптаза-2 (matriptase-2), которую кодирует ген TMPRSS6, и большой мультифункциональный трансмембранный протеин неогенин (neogenin). Неогенин регулирует гомеостаз железа путем угнетения секреции HJV, ингибирующего сигнализацию BMP, что, в свою очередь, приводит к повышению экспрессии гепсидина. Матриптаза-2 является трансмембранной сериновой протеазой, оказывающей негативное влияние на продукцию гепсидина, путем расщепления гемоювелина [60,129,163,221,256].
Эритропоэтическая активность играет важную роль в регуляции продукции гепсидина. Уровень гепсидина снижается при железодефицитной анемии, гемолитических анемиях и анемиях с неэффективным эритропоэзом. Однако механизм снижения продукции гепсидина при этих состояниях может быть различным. Эти механизмы могут включать в себя растворимые протеины, вырабатывающиеся в костном мозге эритробластами, снижение циркулирующего железа или его запасов и гипоксии. Молекулярные основы, вовлеченные в данные механизмы, пока остаются неясны. Эритропоэтин (ЭПО) индуцированный гипоксией может действовать как посредник и угнетать продукцию гепсидина
через ЭПО-стимулированную пролиферацию эритробластов, продукты секреции предшественников эритроцитов и, в конечном счете, повышение утилизации железа для синтеза гемоглобина [99,195,210].
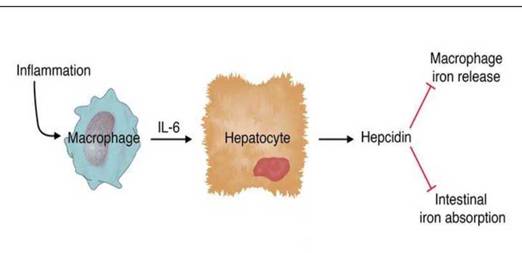
Воспаление играет ведущую роль в регуляции синтеза гепсидина. Экспрессия гепсидина индуцируется липополисахаридами и интерлейкином-6, и ингибируется ФНО-а [12,107]. Синтез гепсидина быстро повышается при инфекции и воспалении. ИЛ-6 является основным индуктором гепсидина и действует через STAT3-зависимый транскрипционный механизм. У волонтеров, которым был введен ИЛ-6, экскреция гепсидина с мочой увеличилась в несколько раз уже через 2 часа после инфузии (Рисунок 1.3.) [89,92,95,106,213,239].
Рисунок 1.3. Роль воспаления в регуляции синтеза гепсидина [89]
У мышей, которым вводились провоспалительные цитокины - интерлейкин-1 и фактор некроза опухолей а (ФНО-а) развились снижение содержания железа в сыворотке и анемия [114]; комбинация этих состояний была связана с цитокин- индуцированным синтезом ферритина - главного белка, ассоциированного с хранением железа макрофагами и гепатоцитами [225]. При хроническом воспалении поглощение железа макрофагами происходит путем эритрофагоцитоза и трансмембранным импортом с участием белка - двухвалентного металлотранспортера 1 (ДМТ1) [13,130].
ИФН-y,липополисахариды, ФНО-а повышают экспрессию ДМТ1, который в свою очередь повышает поглощение железа активированными макрофагами [13]. Эти провоспалительные стимулы также индуцируют снижение экспрессии ферропортина, таким образом, блокируя выход железа из макрофагов и его накопление в них [43]. Ферропортин - трансмембранный экспортер железа, как полагают ответственный за транспорт адсорбированного «пищевого» железа из энтероцитов в циркуляцию [179]. Более того, противовоспалительные цитокины, такие как ИЛ-10, могут вызывать анемию, через стимуляцию трансферрин- индуцированного поглощения железа макрофагами и трансляционной стимуляции экспрессии ферритина [197].
Снижение продукции эритропоэтина
Эритропоэтин централизованно регулирует пролиферацию эритроидных клеток. Экспрессия эритропоэтина обратно зависит от оксигенации тканей и уровня гемоглобина. Продукция эритропоэтина у пациентов с АХБ не соответствует степени анемии в большинстве, но не во всех случаях [46,47].
Цитокины - ИЛ-1 и ФНО-а напрямую ингибируют экспрессию эритропоэтина in vitro - что вероятно объясняется, по крайней мере частично, цитокин- индуцированным образованием активных форм кислорода, которые в свою очередь приводят к повреждению эритропоэтин-продуцирующих клеток. Хотя убедительных данных, полученных в исследованиях на людях недостаточно, инъекции липополисахаридов мышам приводят к редукции экспрессии мРНК эритропоэтина в почках и снижению уровня циркулирующего эритропоэтина [134].
Чувствительность клеток-предшественников к эритропоэтину, вероятно, обратно связана с основными причинами хронического заболевания и количеством циркулирующих в крови цитокинов. Концентрация интерферона-Y или ФНО-а гораздо выше количества эритропоэтина, необходимого для восстановления образования Э-БОЕ [8,150].
Чувствительность к эритропоэтину еще больше снижается посредством ингибиторных эффектов провоспалительных цитокинов, направленных на пролиферацию эритроидных клеток-предшественниц. Параллельно снижение рецепторов эритропоэтина и ограничение доступности железа вносят свой вклад в замедление пролиферации и синтеза гемоглобина. Наконец, повышенный эритрофагоцитоз приводит к снижению продолжительности жизни эритроцитов, наряду с предполагаемым повреждением эритроцитов цитокинами и свободными радикалами [31,207].
Замедление пролиферации эритроидных клеток-предшественников
У пациентов с АХБ пролиферация и дифференцировка эритроидных предшественников - эритроидных бурстобразующих единиц (Э-БОЕ) и эритроидных колониеобразующих единиц (Э-КОЕ) - замедлена и связана с ингибиторным эффектом интерферона-а, -в и -у, ФНО-а, интерлейкина-1, которые влияют на рост Э-БОЕ и Э-КОЕ [151]. Интерферон-Y видится наиболее мощным ингибитором, что отражено обратной корреляцией между его уровнем в крови и концентрацией гемоглобина и числа ретикулоцитов [241]. Основной механизм может включать цитокин-опосредованную индукцию апоптоза, который, возможно, частично связан с образованием церамида и снижением экспрессии рецепторов эритропоэтина на клетках-предшественницах, угнетением образования эритропоэтина и снижение экспрессии других прогемопоэтических факторов, таких как фактор стволовых клеток [151,119,241]. Кроме того, цитокины оказывают прямое токсическое воздействие на клетки- предшественники, вызывая образование лабильных свободных радикалов, таких как окись азота или супероксид анион, расположенными рядом макрофагами [167].
Кроме того, было описано, что гепсидин угнетает эритропоэз (in vitro) при сниженной концентрации эритропоэтина [44]. А нарушение гомеостаза железа, вследствие гиперпродукции гепсидина, приводящее к ограничению доступности
железа для эритроидных клеток-предшественников приводит к замедлению их деления вследствие негативного влияния на биосинтез гемма [245].
1.2.2.
Еще по теме Основы патогенеза анемии при хронических болезнях:
- Варианты терапии анемии при хронических болезнях и их эффективность
- Диагностика анемии при хронических заболеваниях
- 1.2 Анемия при хронических болезнях
- VIII. 1.2. Прогредиентность и хронический характер наследственных болезней
- VII.3.2. Особенности патогенеза хромосомных болезней
- 1.3. Сочетание хронической обструктнвной болезни легких н бронхиальной астмы
- VII.3.1. Особенности патогенеза моногенных болезней
- Особенности патогенеза ожоговой болезни глаз
- Личность в условиях болезни. Варианты личностного развития в условиях хронического соматического заболевания.
- Патология иммунного надзора как основа патогенеза ревматических заболеваний
- Хронический кандидоз кожи и слизистых оболочек (хронический генерализованный гранулематозный кандидоз, эндокринно-кандидозный синдром, кандида-гранулема)
- Клинико-лабораторные показатели при лечении хронического артрита с использованием биологической терапии
- Приложение 1 Паллиативная помощь и уход. Медицинские, психологические и духовные аспекты при ВИЧ/СПИДе и прогрессирующих хронических заболеваниях
- XI.1.2. Умственная отсталость при моногенных болезнях
- Медицинские, психологические и духовные аспекты при ВИЧ/СПИДе и прогрессирующих хронических заболеваниях ОПЫТ РУССКОЙ ПРАВОСЛАВНОЙ ЦЕРКВИ