Х.1.1. Аутосомно-доминантные заболевания
Примером аутосомно-доминантного заболевания может служить синдром Марфана, для которого характерны высокая пенетрантность и различная экспрессивность.
Частота его составляет 1:10 000. Синдром Марфана, впервые описанный в 1886 г., представляет собой генерализованное поражение соединительной ткани. В настоящее время известно, что в основе болезни лежит мутация в гене фибриллина — белка, входящего в состав соединительной ткани и обеспечивающего ее упругость. Ген локализован на 15-й хромосоме в участках 15q21.1. В клинической картине выделяется поражение трех систем организма: опорно-двига-тельной, сердечно-сосудистой и органов зрения.Все больные имеют характерный внешний вид: высокий рост, астеническое телосложение.
Нарушения опорно-двигательной системы включают непропорционально длинные пальцы (арахнодактилия, или «паучьи» пальцы), долихоцефальный череп, деформацию грудной клетки (воронкообразная или килевидная), искривление позвоночника (сколиоз, кифоз), гиперподвижность суставов, плоскостопие (рис. X.1). Со стороны сердечно-сосудистой системы наиболее характерны пролапс митрального клапана, расширение аорты в восходящем или брюшном отделе с развитием аневризмы. Патология органов зрения в виде миопии высокой степени связана с подвывихом (или смещением) хрусталика, гетерохрония (разный цвег) радужки. Нередко отмечаются паховые, бедренные, диафрагмальные грыжи. В редких случаях описаны опущение почек, эмфизема легких, тугоухость и глухота. Психическое и умственное развитие больных соответствует норме. Прогноз для жизни и продолжительность жизни определяются степенью поражения сердечно-сосудистой системы.
Синдром Холт-Орама (синдром рука—сердце) представляет собой моногенный синдром множественных врожденных пороков развития.
Клиническая картина характеризуется аномалиями верхних конечностей и врожденными пороками сердца. Пороки развития руки варьируют от недоразвития или отсутствия 1-го пальца кисти и трехфалангового 1-го пальца кисти до недоразвития или полного отсутствия лучевой кости с формированием лучевой косорукости. Чаще поражается левая рука. Наблюдаются и другие скелетные изменения: гипоплазия лопаток и ключиц, сколиоз, воронкообразная деформация грудины, искривление мизинца, сросшиеся пальцы (синдактилия), гипоплазия других пальцев кисти.
В 85% случаев у больных обнаруживаются различные формы врожденных пороков сердца: дефекты межпредсердной и межжелудочковой перегородок, открытый аортальный проток, коарктация аорты, стеноз легочной артерии, пролапс митрального клапана и др.
В связи с варьирующей экспрессивностью синдрома и с целью выявления минимальных проявлений патологического гена родственники больных должны быть тщательно осмотрены и обследованы (рентгенография кисти и эхокардиография). Диагноз ставится на основании клинико-генеалогических данных и параклинического обследования. Прогноз жизни зависит от тяжести поражения сердца.
Еще по теме Х.1.1. Аутосомно-доминантные заболевания:
- 1Х.4.1. Аутосомно-доминантный тип наследования заболевания
- Изолированная аутосомно-доминантная атрофия зрительного нерва (вариант Kjer)
- Аутосомно-доминантная атрофия зрительного нерва, сочетающаяся с глухотой, офтальмоплегией, дистаксией и миопатией
- Аутосомно-доминантная атрофия зрительного нерва, сочетающаяся с врожденной глухотой
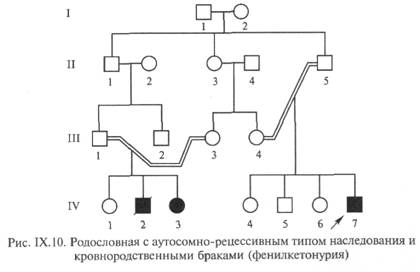
- IХ.4.2. Аутосомно-рецессивный тип наследования заболевания
- Доминантный Х-сцепленный тип наследования заболевания
- Х.1.2. Аутосомно-рецессивные заболевания
- 9. Генетические заболевания. Хромосомные аномалии. Аутосомные и сцепленные с полом признаки.
- доминантная атрофия зрительного нерва
- Моногенные болезни, наследуемые по аутосомно–доминантному типу
- Доминантные лексические категоризации говорения и их концептуализация в литературном языке и в диалекте
- VIII.1.4. Семейный характер наследственных заболеваний
- Факультативные и облигатные предраковые заболевания
- 5.4 Заболевания органов пищеварения.
- Х.1.3. Х-сцепленные рецессивные заболевания
- 70 Соматогенные психические заболевания
- Эндокринные заболевания
- Заболевания вегетативной нервной системы .
- Заболевания тонкого и толстого кишечника.