Х.1.2. Аутосомно-рецессивные заболевания
Среди аутосомно-рецессивных заболеваний наиболее распросӯтраненным является муковисцидоз (кистофиброз поджелудочной железы).
Частота его среди новорожденных в европейской популяции составляет 1:2500.
Заболевание обусловлено генерализованным поӯражением экзокринных желез. Ген муковисцидоза расположен на 7-й хромосоме. Этот ген контролирует синтез белка, называемого трансмембранным регулятором проводимости.Патогенез болезни обусловлен тем, что при отсутствии синтеза трансмембранного регулятора (первичного продукта гена) наруӯшается транспорт хлоридов в эпителиальных клетках. Это привоӯдит к избыточному выведению хлоридов, следствием чего являетӯся гиперсекреция густой слизи в клетках поджелудочной железы, бронхов, слизистой оболочке желудочно-кишечного тракта. Выӯводные протоки поджелудочной железы закупориваются, слизь не выводится, образуются кисты. Ферменты поджелудочной железы не поступают в просвет кишечника. Гиперпродукция слизи в бронӯхиальном дереве ведет к закупорке мелких бронхов и последующеӯму присоединению инфекции. Подобные процессы развиваются в придаточных пазухах носа и в канальцах семенников. В потовой жидкости повышена концентрация ионов натрия и хлора, что и является основным диагностическим лабораторным тестом.
Различают следующие клинические формы муковисцидоза: смеӯшанная (легочно-кишечная — отмечается у 65-75% больных); преимущественно легочная (у 15-20%); преимущественно кишечӯная (у 5-10%); мекониальный илеус (не более чем у 1%); стерӯтые и абортивные формы (незначительная доля).
На практике наиболее часто приходится сталкиваться со смеӯшанными формами. Первые симптомы заболевания появляются на первом году жизни, как правило, на фоне перенесенной острой респираторной вирусной инфекции и характеризуются приступоӯобразным навязчивым кашлем, явлениями обструкции и воспалеӯния в легких. Рецидивирующий хронический инфекнионно-воспалительный процесс осложняется гнойно-обсгруктивным бронӯхитом, пневмониями, возникающими несколько раз в год.
К вторичным изменениям относятся бронхоэктазы, эмфизема, пнев-москлсроз, легочное сердце. Параллельно у детей отмечается симӯптоматика со стороны желудочно-кишечного тракта. Нарушения пищеварения проявляются недостаточной прибавкой массы тела, вздутием живота, обильным зловонным стулом с примесью жира. Аппетит у детей сохраняется. В дальнейшем в патологический проӯцесс вовлекается печень (жировая инфильтрация, холестатический гепатит, цирроз).| Мекониальный илеус — это врожденная форма заболевания, проявляющаяся в первые сутки после рождения отсутствием отхождения мекония и клиникой полной кишечной непроходимости. Интеллектуальное развитие у детей не страдает. Прогноз жизни при всех формах заболевания неблагоприятный. В настоящее время пациенты со смешанными формами редко живут более 20 лет. Для заболеваний с аутосомно-рецессивным типом наследования нередко характерно тяжелое течение с поражением различных систем органов и развитием комбинированӯных дефектов. Так, сочетанное наӯрушение умственного развития и зрения наблюдается при синдроме Барде-Бидля. Для синдрома харакӯтерны пять признаков: ожирение, гипогенитализм, умственная отстаӯлость, пигментная дегенерация сетӯчатки и полидикталия (рис. Х.2). Наиболее часто встречающимся (до 90 % случаев) является прогрессиӯрующая пигментная дегенерация сетчатки глаза, приводящая к поӯтере зрения. К 20 годам 75% больӯных становятся слепыми. Из других нарушений зрения описаны катаӯракта, атрофия зрительных нервов, нистагм. У 80% больных уже на первом году жизни развивается ожирение, которое с возрастом прогрессирует. Почти у всех больных с синдромом Барде-Бидля отмечается умственӯная отсталость, а в 75% случае шеӯстипалость (полидактилия) со стоӯроны 5-го пальца. Нередко у больӯных наблюдается гипогонадизм, проявляющийся у мальчиков недоразвитием наружных половых орӯганов, крипторхизмом или гипоспадией, а у больных девочек - недоразвитием яичников, пороками развития матки и влагалища. Характерным для синдрома является разнообразная патология поӯчек. | 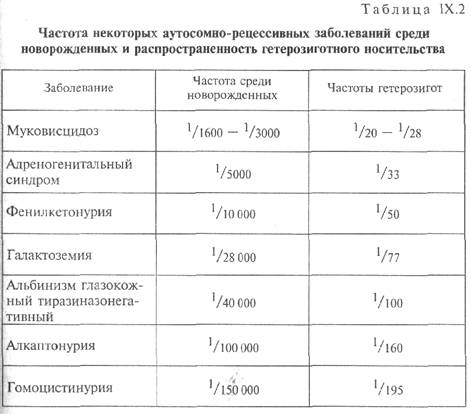 |
Еще по теме Х.1.2. Аутосомно-рецессивные заболевания:
- IХ.4.2. Аутосомно-рецессивный тип наследования заболевания
- Рецессивный Х-сцепленный тип наследования заболевания
- 1Х.4.1. Аутосомно-доминантный тип наследования заболевания
- Х.1.1. Аутосомно-доминантные заболевания
- Х.1.3. Х-сцепленные рецессивные заболевания
- 9. Генетические заболевания. Хромосомные аномалии. Аутосомные и сцепленные с полом признаки.
- Изолированная аутосомно-доминантная атрофия зрительного нерва (вариант Kjer)
- Моногенные болезни, наследуемые по аутосомно–рецессивному типу
- Глава 117 Рецессивная диатеза с нулевым маркером
- Аутосомно-доминантная атрофия зрительного нерва, сочетающаяся с глухотой, офтальмоплегией, дистаксией и миопатией
- Моногенные болезни, имеющие сцепленный с полом рецессивный тип наследования
- Глава 115 Рецессивная диатеза и маркер рефлексивности
- Аутосомно-доминантная атрофия зрительного нерва, сочетающаяся с врожденной глухотой
- Глава 1 1 6 Рецессивная диатеза с маркером пассива