СИНДРОМ ОЛДРИЧА (синдром Олдрича — Ннскотта)
В 1954 г. Aldrich с соавт. описал синдром, при котором у детей раннего возраста имеется тромбоцитопеническая пурпура, экзема, гнойный отит и кровавый понос. Вслед за этим последовали другие сообщения (Roberts, Smith, Schaar), подтвердившие существова-
I (14

ждениыми.
Наиболее редко встречается врожденная гипопластическая форма амегакариоцитарной пурпуры. Эта форма тромбоцитопении относительно часто сочетается с другими врожденными уродствами. Одно из первых описаний этого заболевания принадлежит Greenwald и Sherman. У больных с гипопластической амегакариоцитарной тромбоцитопенией наблюдаются нередко костные аномалии. Заслуживает внимания, что гипопластическая тромбоцитопения иногда сочетается с врожденным отсутствием лучевой кости. Это объясняется тем, что внутриутробное развитие мегакариоцитарногО аппарата и лучевой кости происходит в одно и то же время (6—8 недель). Врожденные амегакариоцитарные гипопластические тромбоцитопении встречаются реже, чем врожденные гипопластические анемии, при которых имеется поражение миелоидных, эритроидных и мегакариоцитарпых элементов костного мозга. В клинической картине гипопластических тромбоцитопений повышенная кровоточивость может проявляться сразу после рождения или несколько позже, в раннем грудном возрасте. Если кровотечения при гипопластической тромбоцитопении появляются вскоре после рождения, у больных может развиться лейкемоидная реакция (Emery с соавт.).В литературе имеются описания амегакариоцптоза с костными деформациями у детей из двойни, что свидетельствует о генетической природе этого редкого заболевания.
Гормональное лечение (ΛΚΤΓ, предилзолон) обычно эффекта не дает. При полном отсутствии мегакариоцитов в костном мозгу спленэктомия противопоказана, при значительном снижении мегакариоцитов эта операция дает лишь временный гемостатический эффект.
Прогноз неблагоприятный, большинство больных погибает в первые недели или месяцы жизни, несмотря па интенсивную терапию (трансфузии крови, гормональное лечение, спленэктомия). По данным Stefanini и Daineshcli1 продолжительность жизни детей с врожденной амегакариоцитарной тромбоцитопенией н костными деформациями не превышает 3— 4 лет. От врожденной гипопластической тромбоцитопении следует отличать преходящую гипоплазиюкостного мозга с геморрагическими явлениями у недоношенных детей, которая имеет обратимый характер и прогностически — бла-
Iоприятна.
Для иллюстрации приводим собственное наблюдение.
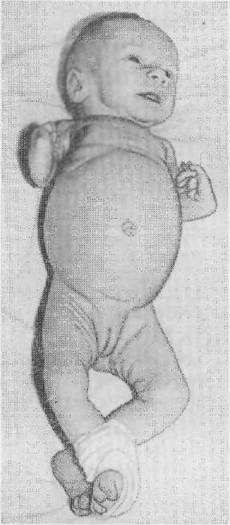
Рис. 7. Де почка Е. IL с амего- карноцнтарной тромбоцитопенией и уродством верхних конечностей.
Больная E1 Н. поступила в 17-ю детскую больницу Ленинграда в возрасте 14 дней. Из анамнеза известно, что она от нерпой беременности молодых еюровых родителей Наследственность не отягощена. Мать,
18 лет, во время беременности ничем не болела, лекарств не принимала. Роды в срок. Депочка родилась в синей асфиксии с весом 3150 г и ростом 50 см. При рождении обнаружено уродство верхних конечностей: отсутствие .лучевых костей, укорочение локтевых костей н косорукость (рис, 7). Направлена в больницу В СВЯЗИ C ухудшением общего состояния, плохой прибавкой веса, частыми срыгиванпями, одышкой н пероральным цианозом, В стационаре диагностированы: врожденное уродство верхних конечностей. двусторонняя мелкоочаговая бронхопневмония, пи- лороспаэм и гипотрофия II степени. В исследовании крови обнаружен лейкоцитоз — 19 SOO с нейтрофилезом—-56% (3/Ш 1971 г.). На рентгенограмме
грудной клетки двусторонняя мелкоочаговая пневмония. На рентгенограммах предплечий определяется отсутствие лучевых костей η косорукость. Неоднократные исследования тромбоцитов в 1971 г. давали тромбоцитопению — от единичных в препарате до 30 000 в
1 Λ.ui, Нейтрофильный лейкоцитоз все время пребывания в стационаре держался от 19 800 до 50 800, РОЭ от 2 до 40 мм в I ч.
Посев крови стерилен. Время свертывания крови 8, по Mac- Al агро. Длительность кровотечения 3', Ретракция кровяного(rycτκn плохо выражена. Эндотелиальные пробы отрицательны. H мнеляграмме мегакариоикгы единичные в препарате, недеятель ные. в остальном картина костного мола в пределах возрастной нормы. На основании миелограммы и упорной тромбоцитопении в клинике установлен диагноз врожденного амегакарноцитоза η сочетании с врожденными костными деформациями. Геморрагические явления у больной проявились в кровотечениях из желудочно-кишечного тракта. За время пребывания η больнице развилась анемия — количество эритроцитов снизилось до 3 IOOO(X) и гемоглобин до 53 ед. Ретнкулоиитоз 5,1%. Общее состояние девочки прогрессивно ухудшалось. Лечение трансфузиями крови, предниэолоном, антибиотиками, е-аминокапроновой кислотой и симптоматическими средствами оказалось не эффективным и больная скончалась η возрасте 3l⅛ месяцев. Секционный диагноз: врожденная амегакариоцнтарная тромбоцитопения в сочетании с пороком развития обеих верхних конечностей (отсутствие лучевых костей, укорочение локтевых, двусторонняя косорукость), Крупноточечные кровоизлияния в слизистую оболочку желудочно- кишечного тракта. Двусторонняя бронхопневмония. Истощение.
Еще по теме СИНДРОМ ОЛДРИЧА (синдром Олдрича — Ннскотта):
- Синдром «вьюнка» (синдром «утреннего цветка» или «ипомеи»)
- Синдром диссеминированного внутрисосудистого свертывания крови — ДВС-синдром.
- (мпогоотвпрстпын прозивний эктодермоз, дерматостоматит, елизнето-кожно-глазнон синдром, синдром Лайлы, п люрпорифициа ль пы й э кто дермоз)
- Синдром Мартина – Белла (синдром ломкой Х–хромосомы).
- Синдром Бехчета, або офтальмостоматогенітальний синдром.
- 48 Абстине?нтный синдро?м у наркоманов
- 13.12. Папиллоренальным синдром
- Психогенна сплутаність (синдром Ганзера)
- Синдром выгорания
- 13.13. Синдром Эйкарди
- Абстинентный синдром
- 40. Синдром похмелья.
- Синдром «кошачьего крика»
- 9.12. Синдром Элерса -Данло