Кристаллизация аморфных Ge2Sb2Te5
Ранее мы исследовали стабильные позиции примеси, сравнивая полные энергии между вероятными местами в c-GST. Для того, чтобы непосредственно определить позиции примеси в C-GST, мы выполняем моделирование отжига легированных a-GST, ожидая перекристаллизации в c-GST.
Это моделирование может быть полезным при изучении влияния примесей на динамику кристаллизации. Для уменьшения вычислительных затрат, 38мы используем решетку из 72 атомов, включая одну SiGe, N⅛, Oj из примесей в элементарную ячейку. При такой относительно небольшой элементарной ячейке, максимальное время моделирования, что возможно с имеющимися вычислительными ресурсами только 1 нс, что на порядок меньше, чем типичное время кристаллизации GST. В работе [56] показано, что сжатие a-GST ускоряет процесс кристаллизации. Используя эту идею, мы изотропно уменьшили объем аморфного GST на 6% и отжигали при температуре 650К (чуть ниже температуры плавления) около 400 пс. Затем происходило охлаждение со скоростью -15 К/пс до температуры 300 К и полной релаксации при 0 К. На рисунке 2.7а показана общая энергия за исключением кинетической энергии ионов. В начале координат установлено значение энергии для каждого из легированных с-GST при 0 К.
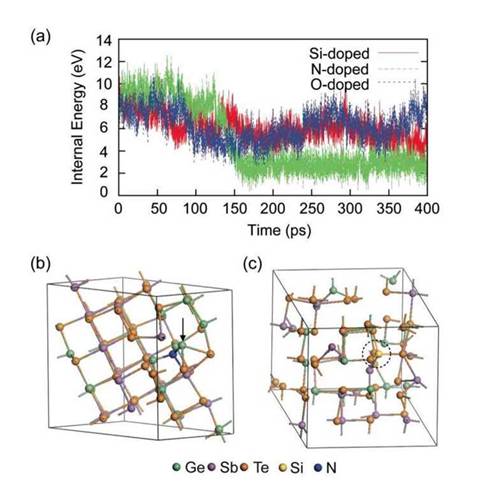
Рис. 2.7. Динамика кристаллизации легированных a-GST: (а) временное изменение внутренней энергии a-GST относительно времени отжига. Температура отжига выбрана 650 К; (б) окончательная структура N-легированного GST после отжига. Дефектный атом Ge указан стрелкой; (в) окончательный состав Si- легированного GST. Атом Si отмечен пунктирной окружностью.
В то время как внутренняя энергия N- легированного GST значительно падает при 130πc, для Si- и О- легированных a-GST изменяется незаметно Конечная установившаяся структура N-легированного GST на рисунке 2.76 подтверждает, что кристаллизация закончена.
Разность энергии между структурой до и после отжига (ДЕ отжига) 4.83 эВ для N-легированного GST. Величина эта значительно меньше, чем ДЕ,м.для нелегированного GST для которого 6,87 эВ при 72 атомов клетки, что указывает на относительную стабильность a-GST и усиливается с добавлением легирующей примеси. Отметим, что полная энергия конечной структуры выше на 1,8 эВ, чем 72 - атомная с-GST. То есть, N- легированный GST кристаллизуется в метастабильное структуры. Это объясняется структурными дефектами индуцированных примесей N. C другой стороны, ЛЕ отжига для Si- легированного GST равна -3,46 эВ для окончательной структуры (из рисунка 8с видна неполная кристаллизация) Несмотря на это, вариации внутренней энергии O- легированных GST аналогичны Si-легированному GST, но ЛЕ отжига равна только 1 эВ, а окончательная структура все еще близка к аморфной. В работе 56 так же под давлением а- GST подвергали перекристаллизации при IOO пс. Время кристаллизации N-легированного GST немного больше, чем это значение. Таким образом, очевидно, что N-легирование с концентрацией 1,4 ат. % недостаточно, чтобы повлиять на динамику кристаллизации а- GST. Запаздывание кристаллизации N-легированного GST может быть в результате кластерных примесей, которые существуют в более высокой концентрации легированияЛегирующей примеси N в кристаллическом GST связан с тремя атомами Ge и одним атомом Sb длинами связей 2,02 и 2,39A соответственно (рис. 2.76). Среди них, два из трех атомов Ge были соединены в начальной аморфной структуре, и они не разрушаются во время процесса рекристаллизации. Атом Ge, указанный стрелкой на рисунке 2.76 не является совместимым по типу строения атомной решетки и вносит значительные деформации решетки вокруг примеси Более конкретно, дефектный атом Ge вращается кристаллографической ориентацией соседних атомов Те, которые можно рассматривать как примитивные формы границы зерна. C другой стороны, соседние атомы Si (три атома Те и один атом Sb) отличаются от соседей в исходной аморфной фазе, и они связаны в процессе отжига.
Это означает, что Si-Te связи слабее, чем Ge-N связи. Si примеси на рисунке 2.7в по-прежнему тетракоординированные, хотя большинство атомов приведены в соответствие со структурой каменной соли. Этот факт означает, что энергетический барьер из тетраэдрических sp3связей с ортогональными р связями образует сеть и имеет существенное значение для атомов Si, который представлял бы собой микроскопическое препятствие кристаллизации Si-легированного GST
Мы провели обширные неэмпирические вычисления кристаллических и аморфных структур GST легированного атомами Si, N, О с целью объяснить выраженное воздействие легирующих примесей на атомном уровне. В кристаллической фазе наиболее благоприятное место для si-легирования это SiGe, в то время как N и О примеси замещают атомы Те из-за сильных связей Ge-N или Ge-O Тем не менее, сравнение параметров решетки с экспериментом показывает, что интерстициальные примеси должны существовать в значительных количествах. Анализ на плотности состояний указывает, что локализация в верхней валентной зоне увеличивается на N- или О-легированных c-GST, что увеличивает сопротивление p-типа c-GST. Аморфные структуры легированных GST были получены моделированием охлаждения расплава. Было обнаружено, что для каждой примеси формируется предпочтительная связь, например Si-Те, Ge-N, и Ge-O В частности, местные структуры связей вокруг атомов азота или кислорода хорошо объясняются на основе кристаллических структур Ge3N4 и GeO2. Это похоже на кристаллическую фазу, но локальная геометрия связей является более благоприятной в аморфной фазе, благодаря которой проявляется повышенная устойчивость легированных a-GST. Анализ локальных структур опроверг точку зрения, что характер связи значительно изменяется на ковалентный. Легирующие примеси N повысили ковалентной характер Ge, но это было компенсировано увеличением металлической природы атомов Те. Мы также проводили моделирование отжига a-GST и атомная структура перекристаллизовывалась в N-легированном GST и была близка к Nj примеси в C- GST.
Тем не менее, сильная тенденция легирующей примеси N образовывать связи с атомами Ge, вызывали серьезные деформации решетки на границе зерна. Легированный кремнием GST была частично кристаллической, но атомы Si остались в тетраэдрической конфигурации Сравнение времени кристаллизации с нелегированным материалом подтвердило, что Si и О примеси замедляют процесс кристаллизации a-GST. Описание поведения примесей, электронной и атомной структуры, свойств материала, для современных теоретических исследований будет служить полезным ориентиром в выборе оптимальных легирующих примесей, чтобы спроектировать специфические свойства GSTРекомендуется легирование не менее 5 ат. %. Это обусловлено тем, что при более низкой концентрации легирующей примеси (от 1,4 ат. % до 5ат. %) не оказывается влияние на параметры кристаллизации. При концентрациях близких к 5 ат. % атомы примеси занимают І - места в кристаллической решетке и оказывают максимальное влияние на свойства материала При дальнейшем увеличении концентрации примеси характеристики материала не изменяются или изменяются крайне незначительно.
2.2.
Еще по теме Кристаллизация аморфных Ge2Sb2Te5:
- Примеси в аморфных Ge2Sb2Te5
- 2.1.5. Электронные структуры легированного аморфного Ge2Sb2Te5
- Горизонты аккумуляции аморфной гидроокиси железа
- Ржаво-охристые пятна аморфной гидроокиси железа
- Выращивание методом направленной кристаллизации
- 4.2. Морфология кристаллов германия и ее связь с кинетикой кристаллизации
- Свободные аморфные новообразования
- Расчет размерных зависимостей температуры плавления и кристаллизации металлических нанокластеров
- О результатах исследования структурных характеристик в металлических нанокластерах при фазовом переходе плавление- кристаллизация
- Экспериментальные исследования плавления и кристаллизации наночастиц
- 4.1. Морфология кристаллов парателлурита и ее связь с кинетикой кристаллизации
- О взаимосвязи размерных зависимостей температур плавления и кристаллизации наночастиц металлов
- Расчет размерных зависимостей теплот плавления и кристаллизации наночастиц металлов
- Приложение. Графики и рисунки, описывающие эволюцию структурных характеристик для нанокластеров алюминия и кобальта в процессе плавления и кристаллизации
- Моделирование плавления и кристаллизации металлических нанокластеров, определение параметров гистерезиса калорических кривых потенциальной части внутренней энергии